Article Text

Abstract
Background The 11p15 region contains two clusters of imprinted genes. Opposite genetic and epigenetic anomalies of this region result in two distinct growth disturbance syndromes: Beckwith-Wiedemann (BWS) and Silver-Russell syndromes (SRS). Cytogenetic rearrangements within this region represent less than 3% of SRS and BWS cases. Among these, 11p15 duplications were infrequently reported and interpretation of their pathogenic effects is complex.
Objectives To report cytogenetic and methylation analyses in a cohort of patients with SRS/BWS carrying 11p15 duplications and establish genotype/phenotype correlations.
Methods From a cohort of patients with SRS/BWS with an abnormal methylation profile (using ASMM-RTQ-PCR), we used SNP-arrays to identify and map the 11p15 duplications. We report 19 new patients with SRS (n=9) and BWS (n=10) carrying de novo or familial 11p15 duplications, which completely or partially span either both telomeric and centromeric domains or only one domain.
Results Large duplications involving one complete domain or both domains are associated with either SRS or BWS, depending on the parental origin of the duplication. Genotype-phenotype correlation studies of partial duplications within the telomeric domain demonstrate the prominent role of IGF2, rather than H19, in the control of growth. Furthermore, it highlights the role of CDKN1C within the centromeric domain and suggests that the expected overexpression of KCNQ1OT1 from the paternal allele (in partial paternal duplications, excluding CDKN1C) does not affect the expression of CDKN1C.
Conclusions The phenotype associated with 11p15 duplications depends on the size, genetic content, parental inheritance and imprinting status. Identification of these rare duplications is crucial for genetic counselling.
- Silver Russell Syndrome
- 11p15 Duplication
- Beckwith-wiedemann Syndrome
- Imprinting Disorder
- Cnv
Statistics from Altmetric.com
Introduction
Fetal growth is a complex physiological process that is finely regulated by genetic, hormonal and environmental factors. Among these factors, the role of the IGF system in the control of fetal growth has been particularly well described. Imprinting is defined as the monoallelic expression of a gene according to its parental origin. Approximately 100 genes, implicated in several physiological processes, including growth, are imprinted in humans.1
In humans, the 11p15 region contains several imprinted genes playing a crucial role in the control of fetal and postnatal growth. Indeed, genetic and epigenetic anomalies of this region are associated with two syndromes characterised by growth disturbances: Beckwith-Wiedemann syndrome (BWS) and Silver-Russell syndrome (SRS).1
BWS (MIM 130650) is characterised by prenatal and postnatal overgrowth, macroglossia, organomegaly, abdominal wall defects, including omphalocele, neonatal hypoglycaemia, ear creases/pits, hemihypertrophy and an increased risk of embryonic tumour development.2 In contrast, SRS (MIM 180860) is characterised by restricted prenatal and postnatal growth, a typical facial appearance with triangular face and relative macrocephaly, hemihypotrophy and failure to thrive.3
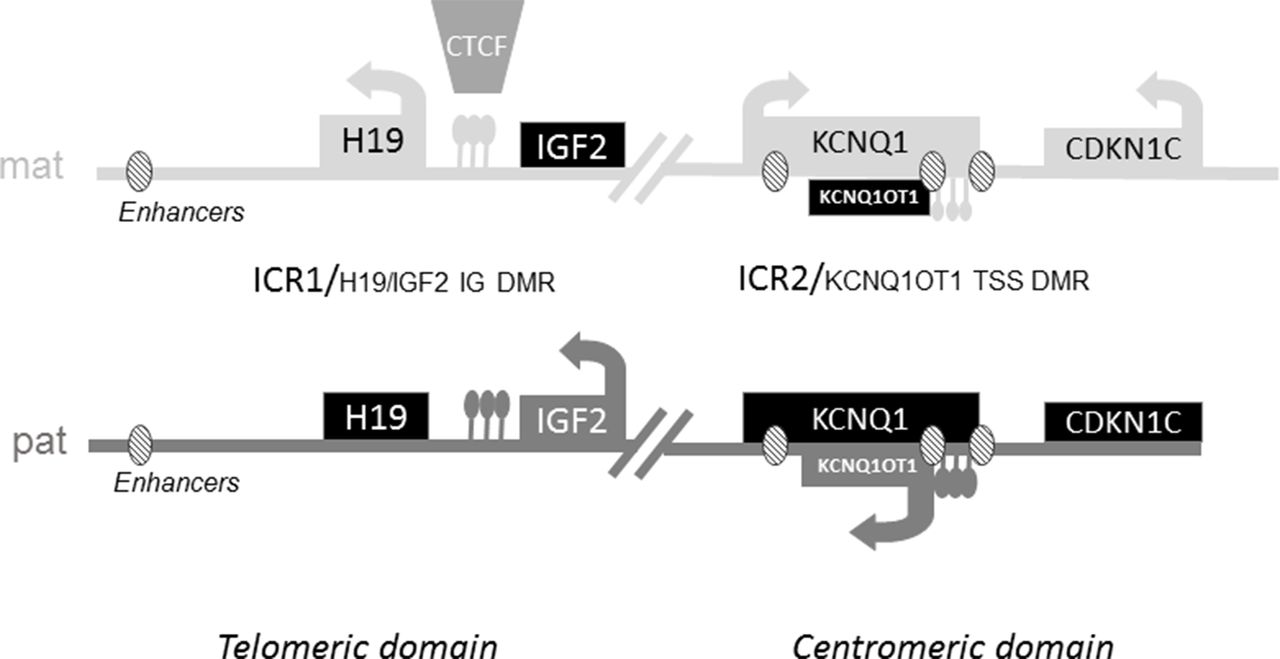
Opposite molecular defects within the 11p15 region result in those two opposing clinical syndromes. The imprinted genes within the 11p15.5 region are clustered in two regulatory domains: one that is centromeric and the other telomeric.4 5 Gene expression is controlled by the methylation status of the two imprinting centres: H19/IGF2:IG DMR (also called ICR1 (Imprinting Control Region 1)) for the telomeric domain and KCNQ1OT1:TSS DMR (also called ICR2) for the centromeric domain (figure 1).6 In addition to the imprinting centres, enhancers located in both domains control the expression of the genes located in the region. Within the telomeric domain, common enhancers shared by IGF2 and H19 are located 5’ of H19.7 Within the centromeric domain, the expression of CDKN1C, which encodes a cyclin-dependent kinase inhibitor and has growth suppressing properties, is controlled by at least three enhancer motifs.8
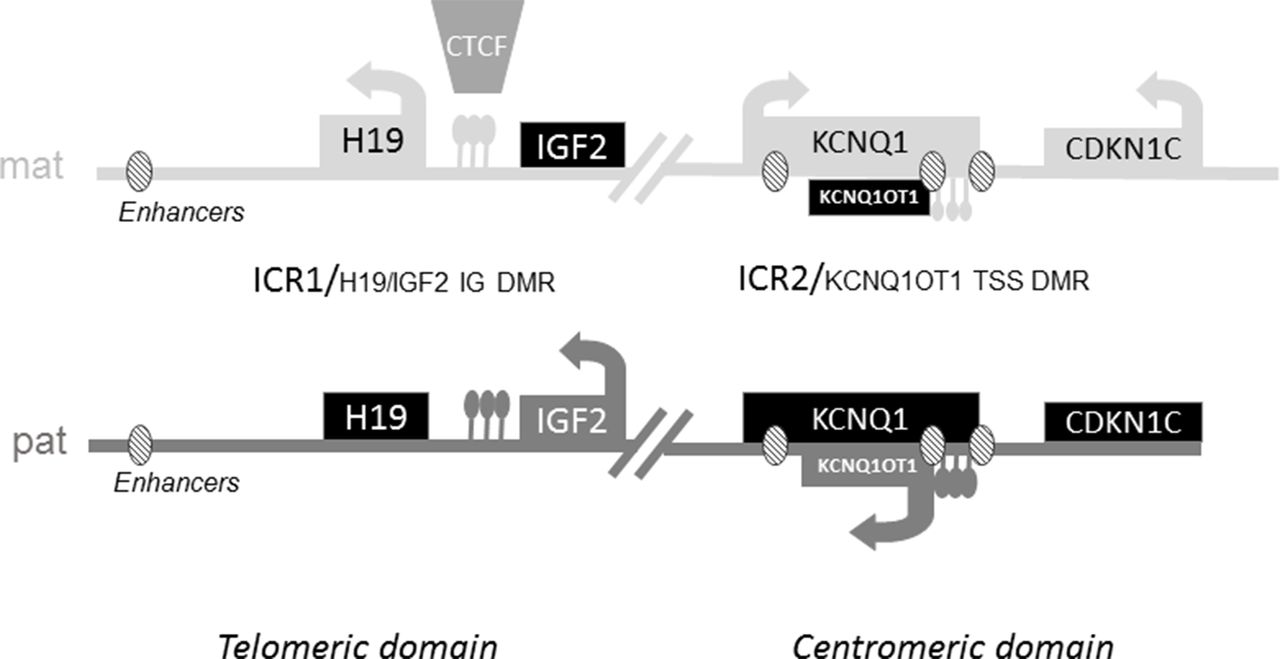
Schematic representation of imprinted gene clusters on 11p15. ICR1 regulates the expression of the non-coding RNA H19 and the IGF2 gene. ICR1 is methylated only on the paternal allele and unmethylated on the maternal allele.6 On the maternal allele, the ubiquitous Zinc-finger DNA-binding protein CTCF can bind to the unmethylated ICR, acting as an insulator and blocking the interaction of downstream enhancers with the IGF2 promoter, leading to transcriptional silencing of IGF2. On the paternal allele, CTCF does not bind to the methylated ICR, allowing the expression of IGF2. In the centromeric domain, ICR2 is methylated on the maternal allele. ICR2 is located in the promoter of the non-coding RNA KCNQ1OT1. The absence of methylation at ICR2 on the paternal allele allows the expression of KCNQ1OT1, which in turn silences all genes of the domain. On the maternal allele, the absence of expression of KCNQ1OT1 allows the expression of Cyclin Dependant Kinase Inhibitor 1C (CDKN1C), an inhibitor of cell proliferation and Potassium Channel Voltage Gated KQT-family number 1 (KCNQ1).6 ICR, Imprinting Control Region.
Loss of methylation (LOM) at the paternal ICR1 is the most frequent molecular defect in SRS, whereas gain of methylation (GOM) at the same locus is observed in 5%–10% of patients with BWS. In contrast, LOM at the maternal ICR2 is observed in approximately 60% of BWS. Epimutation (anticipated to be GOM in SRS) at the same locus has not been observed in SRS. Paternal uniparental disomy (pUPD) of chromosome 11 represents 20% of molecular defects in BWS. In contrast, maternal UPD (mUPD) of chromosome 11 is very rare in SRS,9 whereas mUPD of chromosome 7 is an alternative cause of SRS.10 Finally, mUPD of chromosomes 14, 16 and 20 have also been identified in a few patients with SRS.3 These genetic and epigenetic defects often occur sporadically, with an extremely low risk of recurrence.
In addition to these frequent anomalies, rare genetic defects, such as CDKN1C or IGF2 mutations, can result in a high recurrence risk. Loss of function mutations of maternally expressed CDKN1C leads to BWS,11 whereas a gain of function mutation of this gene has been described in a family with SRS.12 Loss of function mutations of the paternally expressed IGF2 gene and genes which are upstream regulators of IGF2 such as HMGA2 or PLAG1 have also been described.13–16 Moreover, deletions/mutations within ICR1 can be detected in 20% of patients with BWS with ICR1 GOM17 and 1% of patients with SRS with an ICR1 LOM.18
Finally, CNV, including either duplications or deletions within the 11p15 region, are rare and have been identified in less than 3% of patients with SRS and BWS (see online supplementary table 1).19 20 They can be inherited or occur de novo. Large terminal duplications, spanning both domains and derived from unbalanced chromosomal rearrangements, such as unbalanced translocations, have been reported to be associated with SRS or BWS, depending on the parental origin of the duplication.21–25 These large chromosomal anomalies were revealed by karyotyping and subtelomeric FISH. Since the improvement of molecular cytogenetic technologies, such as multiplex ligation-dependent probe amplification (MLPA), array-CGH and SNP-arrays, smaller duplications involving all or part of only one imprinting centre have been found in patients with SRS or BWS.21–30 Interpreting their pathogenic effects is challenging and requires consideration of the clinical features of the patient, along with the mode of inheritance, genetic content and methylation pattern.
Supplementary file 1
Here, we describe 17 new cases of 11p15.5 duplications in patients with SRS and BWS, detected by SNP-array. We compare these variants with those which have been previously reported and discuss their possible functional consequences.
Materials and methods
Patients
All the patients were referred for molecular and cytogenetic analysis for a growth disturbance to a single molecular diagnosis laboratory (Trousseau Hospital, Paris). Each patient has been seen by a clinical geneticist or paediatric endocrinologist and a standardised clinical form was fulfilled by the physician. Their clinical features are shown in tables 1 and 2. All parents signed an appropriate consent form for genetic analysis, in accordance with national ethics rules (Assistance Publique—Hôpitaux de Paris, authorisation n°681 (BWS) and 682 (SRS)). Ten individuals presented with clinical features of BWS (patients 1, 2, 7–12, 18–19) and nine with a clinical diagnosis of SRS (patients 3–6, 13–17).
Clinical features of patients with BWS and 11p15 duplications
Clinical features of patients with SRS and 11p15 duplications
Methylation analysis
Genomic DNA was extracted from peripheral blood lymphocytes using standard procedures.
DNA methylation analysis of the 11p15.5 ICR1 and ICR2 was performed using ASMM-RTQ-PCR (Allele-specific methylated multiplex real-time qPCR) on leucocyte DNA samples, as described elsewhere.31 MLPA was performed using Kit ME030 from MRC Holland (Amsterdam, Netherlands) if methylation analysis showed a methylation abnormality on either ICR1 or ICR2 alone. SNP-array analysis was performed (1) in cases of BWS with an abnormal methylation index for both ICR1 and ICR2 to distinguish between pUPD or duplication of the 11p15.5 region and/or (2) to confirm/characterise the CNV when identified by MLPA.
SNP-array analysis
Index cases and the parents of individuals (when available) were genotyped using cytoSNP-12 or HumanOmniExpress-24 microarrays (Illumina, San Diego, California, USA). Automated Illumina microarray experiments were performed according to the manufacturer’s instructions. Images were acquired using an iScan System (Illumina). Image analysis and automated CNV calling were performed using GenomeStudio V.2011.1 and CNVPartition V.3.1.6. SNP profiles were analysed by examination of signal intensity (Log R ratio, ie, ln (sample copy number/reference copy number)) and allelic composition (ie, B allele frequency).
Karyotyping and characterisation by FISH
Chromosomes were prepared from peripheral blood lymphocyte cultures following standard procedures. FISH with chromosome-specific probes was performed when possible, using probes targeting the H19 and IGF2 locus on the telomeric domain (RP5-998N23 and RP11-889I17, respectively) and probes targeting the KCNQ1, KCNQ1OT1 and CDKN1C locus on the centromeric domain (RP11-116D8, RP4-608B4 and RP11-494F4, respectively).
Results
Nineteen patients with SRS or BWS carried 11p15 duplications that ranged in size from 0.27 to 6.4 Mb (four de novo, nine inherited and six of unknown inheritance). Cytogenetic and methylation data for each patient are shown in table 3. Methylation indices at ICR1 were increased or decreased for duplications present on the paternal or maternal allele, respectively. Conversely, methylation indices at ICR2 were decreased or increased for duplications on the paternal or maternal allele, respectively (except for partial ICR2 duplications in patients 18 and 19).
Cytogenetic and methylation data in patients with SRS/BWS carrying 11p15 duplications
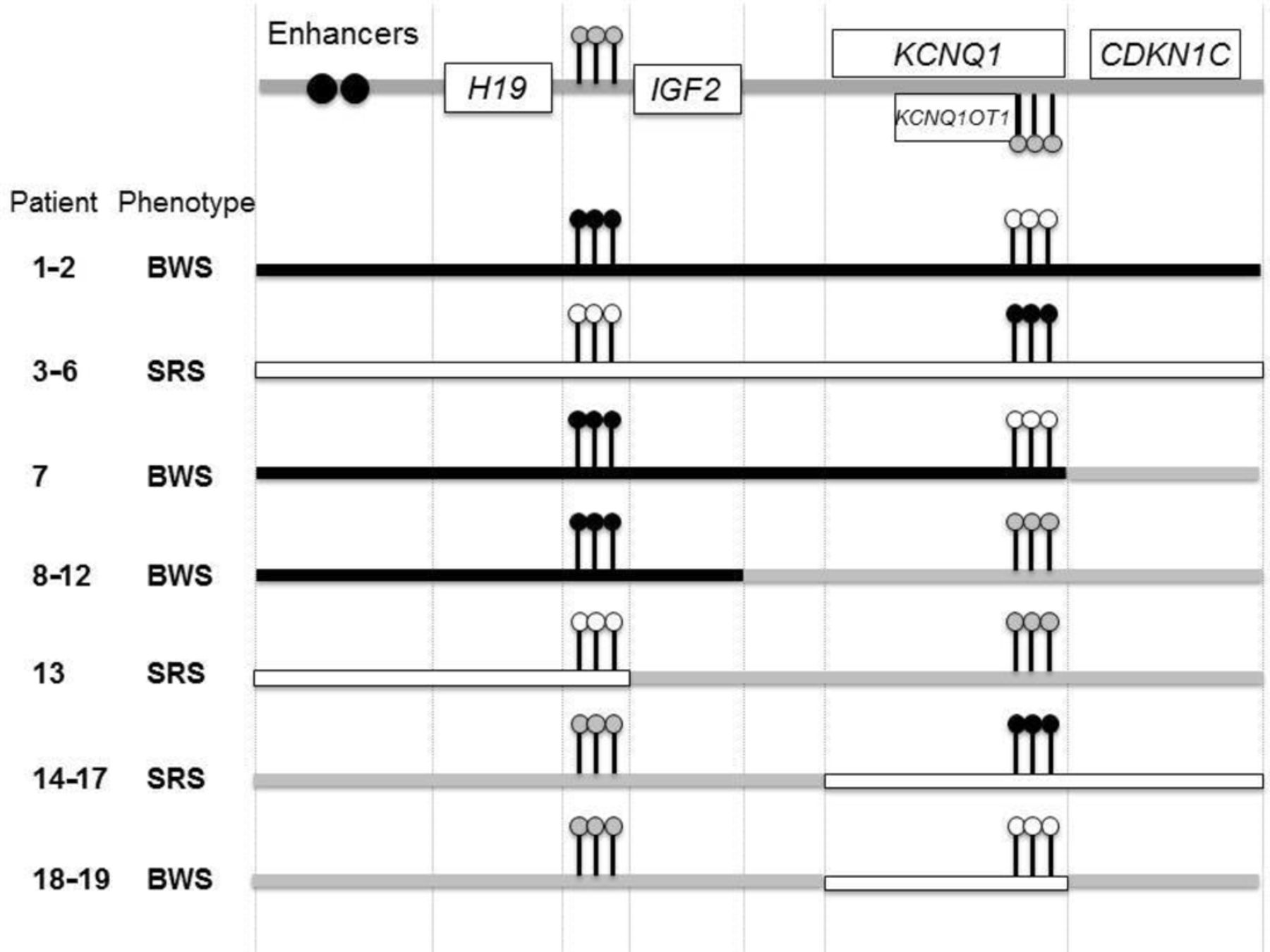
Six patients (patients 1–6) had 11p15 duplications from 1.4 MB to 6.4 MB spanning both domains. Among these, FISH and karyotyping showed that five were unbalanced derivative chromosomes (for example, patient 4, figure 2B) and one corresponded to an interstitial duplication. The 11p15 duplication involving both domains was associated with BWS (patients 1 and 2) when it was of paternal origin, whereas it was associated with SRS (patients 3–6) when it was of maternal origin. Associated deleted segments should be considered to be benign variants, either because of their small size and/or polymorphic content, except for patient 5 for whom the size of the deleted associated segment was 6.8 Mb on chromosome 8p. None of these patients presented with body asymmetry, in accordance with a germinal anomaly instead of a mosaic somatic epigenetic defect. Besides the classical clinical presentation of SRS, patients 5 and 6 presented with salt wasting during early life, requiring fludrocortisone treatment because of mineralocorticoid insufficiency (see section ‘Discussion’).
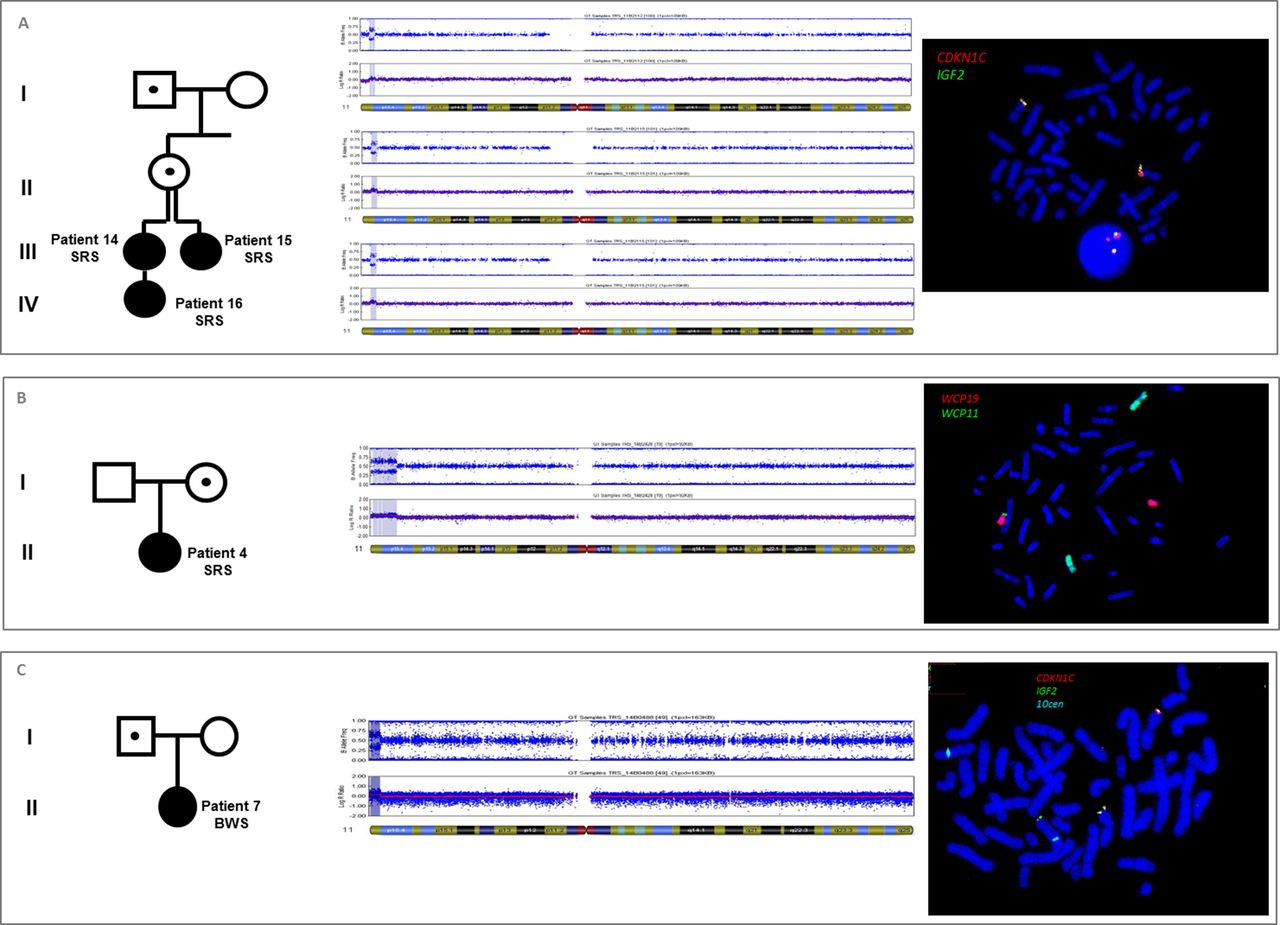
SNP-array and FISH analysis showing 11p15 duplications. (A) 11p15 duplication spanning the whole centromeric domain in patients 14, 15 and 16 with SRS. FISH analysis demonstrated that the 1.4 Mb duplication was located on chromosome 11p. (B) 11p15 duplication of 6.4 Mb spanning both domains, in patient 4 with SRS. FISH analysis with WCP revealed a derivative chromosome 19 (WCP19 red and WCP11 green), carrying the 11p15 duplication. The mother of the patient 4 carried a non-reciprocal balanced translocation t(11;19). (C) 11p15 duplication of 2.5 Mb in patient 7 with BWS. This duplication involved the whole telomeric domain (IGF2 in green) and a part of the centromeric domain, without CDKN1C (in red). FISH analysis showed that the 11p15 duplication was located on a derivative chromosome 10 (centromere of 10 in blue). The father of patient 7 carried the same derivative chromosome 10, but with no clinical effect. BWS, Beckwith-Wiedemann syndrome; SRS, Silver-Russell syndrome; WCP, whole chromosome painting probe.
Patient 7 (figure 2C) had BWS and carried a 2.5 Mb duplication of the entire telomeric domain and a part of the centromeric domain, excluding CDKN1C, coming from an unbalanced translocation t(10;11). Her father carried the same unbalanced translocation, but with no obvious growth disorder phenotype, because of its location on the maternal allele (as the ICR1 and ICR2 methylation indices were decreased and increased, respectively). The associated 10q deletion was a benign variant.
Patients 8–12 had BWS associated with duplication of the entire telomeric domain on the paternal allele and methylation analysis showed an ICR1 GOM and normal methylation of ICR2. All were likely interstitial duplications, except for patient 8 who carried an unbalanced Y derivative chromosome. However, this could not be confirmed because blood samples for FISH analysis were not available for these four patients.
Patient 13 presented with a clinical suspicion of SRS, but with atypical facial dysmorphism, associated with a partial duplication of the telomeric domain (including H19 and ICR1, but not IGF2) of maternal origin. He also carried a 19qter duplication (breakpoints (hg19): 55 650 988–59 097 160) of 3.5 Mb, which may explain the uncommon phenotype.32
Four patients (patients 14–17) carried duplications of the entire centromeric domain, including a familial interstitial duplication segregating through four generations (patients 14–16, figure 2a). In this family, the index cases (patients 14 and 15) were two young half-sisters with SRS and an 11p15 duplication of 1.4 Mb. The two patients inherited the duplication from their mother, who inherited it from her father. The mother and the grandfather showed no features of SRS or BWS. Patient 16 was the daughter of patient 14 and inherited the duplication on the maternal allele and had a SRS phenotype.
Patients 18 and 19 presented with BWS and carried a partial duplication of the centromeric domain (excluding CDKN1C) associated with an ICR2 LOM. Both duplications involved ICR2, part of the KCNQ1 gene (from exons 2 to 11 in patient 18 and exons 11 to 16 in patient 19) and all or part of the KCNQ1OT1 gene (patients 18 and 19, respectively). These were responsible for the BWS phenotype and were associated with the ICR2 LOM. Samples for FISH analyses were not available. Thus, the possibility of an insertion on another chromosome cannot be ruled out. However, tandem duplications are more probable, given the size of the duplicated segment.
Discussion
BWS and SRS mostly occur sporadically, but some patients carry rare genetic variants, exposing them to a high risk of recurrence. We report 17 new familial or de novo 11p15.5 duplications associated with SRS or BWS. 11p15.5 duplications are rare in both BWS and SRS. Small deletions of the same region have been already, but infrequently reported.23 33 Characterisation of these rare molecular anomalies is important for describing the putative functional consequences of the duplications to explain the associated phenotypes and provide accurate information for genetic counselling (figure 3 and see online supplementary table 1 and supplementary figure 1). Duplications of both telomeric and centromeric domains are quite well understood, in which a gene dosage effect of IGF2/CDKN1C explains the growth disturbance. In contrast, partial duplications of one of the domains are more difficult to interpret.
Supplementary file 2

{kind=link}
{kind=link}
![[SP2.jpg]](https://jmg.bmj.com/content/jmedgenet/55/3/205/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
Schematic representation of the reported 11p15 duplications with associated phenotypes and methylation profiles. black boxes: duplication of the paternal allele; white boxes: duplication of the maternal allele. Black lollipops: gain of methylation; white lollipops: loss of methylation, grey lollipops: normal methylation. BWS, Beckwith-Wiedemann syndrome; SRS, Silver-Russell syndrome
Rearrangements within the telomeric domain
IGF2 is a factor which strongly stimulates fetal growth.34 35 Because it is maternally imprinted (paternally expressed), duplications of paternal origin of the whole telomeric domain (as observed in patients 1, 2, 7 and 8–12) lead to overgrowth.21 23 24 36 37 The role of the H19 gene in the control of growth is less clear. Duplication of the whole telomeric domain, which should theoretically be associated with an increased dosage of H19, does not lead to growth retardation when maternally transmitted.21 Only two patients have been identified with partial ICR1 duplications involving only the H19 gene.23 28 In this study, we also describe a patient (patient 13) with an H19 duplication. However, this patient carried another cytogenetic anomaly, as the patient described by Begemann et al,23 which could explain or contribute to his growth restriction phenotype (a 19qter duplication of 3.5 Mb in our patient and a mUPD of chromosome 6 in the patient described by Begemann et al 23).
Other important regulatory elements are the common enhancers located downstream of H19. Deletions of the enhancers on the paternal allele lead to a SRS phenotype,23 38 whereas no case of BWS has been linked to maternal deletions, which should be associated with a decreased dosage of H19. Furthermore, one case of disruption between the enhancers and H19 and IGF2, due to a balanced translocation, has been reported for a patient with SRS with paternal transmission.38 This should lead to decreased expression of H19 on maternal transmission. However, no phenotype was observed in this case. Taken together, these molecular defects favour a more important role for IGF2 rather than H19 in the control of growth, even if a role for H19 cannot be completely excluded.
Rearrangements within the centromeric domain
The regulation of imprinted gene expression for those located on the centromeric domain is less well understood. ICR2 is located in the promoter of the non-coding RNA KCNQ1OT1. Expression of the paternal allele of this gene is required for silencing CDKN1C expression on the same allele. As a cell cycle inhibitor, paternally imprinted/maternally expressed CDKN1C tends to restrain fetal growth.39 Thus, duplications of the whole centromeric domain lead to increased expression of CDKN1C and therefore growth restriction when maternally transmitted, whereas it has no impact on growth when it occurs on the paternal allele (patients 3–6 and 14–17 and references27 30). Furthermore, two patients with SRS carrying such duplications (patients 5 and 6) showed salt wasting due to adrenal insufficiency, likely due to excessive dosage of CDKN1C. This is in accordance with recent descriptions of patients with the rare IMAGe syndrome, caused by CDKN1C gain-of-function mutations.40 41
Duplications of the centromeric domain from the paternal allele should theoretically lead to KCNQ1OT1 overexpression. Despite the fact that we were not able to study genes’ expression in tissues from these patients, the absence of a pathological phenotype on paternal transmission of such duplications (as observed for the mother of patients 14–15 and the mother of patient 17) suggests that KCNQ1OT1 overexpression from the paternal allele does not affect the expression of CDKN1C on the maternal allele, which would theoretically lead to BWS.
The mechanisms that lead to growth disorders due to partial duplications are less clear. We showed that the presence of CDKN1C in the duplicated region is required for growth retardation on maternal transmission. Indeed, the unaffected father of patient 7 carried a large duplication located on the maternal allele, which spanned part of the centromeric domain, excluding CDKN1C. For the father of patient 7, despite ‘hypomethylation’ of IC1 (which is the consequence of a 2:1 ratio between the unmethylated duplicated maternal allele and the methylated non-duplicated paternal allele), the absence of SRS phenotype is probably explained by the normal expression of IGF2 from the normal paternal allele. Conversely, patients 3–6 and 14–17, with duplication of the whole centromeric domain, had SRS because of the maternal origin of the duplication, which includes CDKN1C.
The associated phenotypes in these cases probably all result from a direct dosage effect of CDKN1C from the duplicated allele.
In contrast, partial duplications within the centromeric domain (including the imprinting centre, a part of KCNQ1 and KCNQ1OT1), located on the maternal allele in patients 18 and 19 with BWS, could not be explained by a dosage effect of CDKN1C from the maternal allele. We assume that such duplications lead to the BWS phenotype in those two patients because such partial duplications lead to hypomethylation of ICR2, although excess methylation of the region (due to the gain of maternal chromosomic material) would be expected. This is in accordance with two similar previously reported partial duplications, which led to hypomethylation and BWS on maternal transmission.22 28 Therefore, these duplications within the centromeric domain probably lead to an imprinting defect of the region. Indeed, regulation of CDKN1C on the maternal allele partially depends on enhancer elements which are located throughout this domain8 as well as on the spatial conformation of the chromatin, with allele-specific loops depending on the methylation status of the allele. These loops allow specific interactions between gene promoters and regulatory elements.42 Thus, partial duplications within the centromeric domain may disrupt the regulation of methylation and/or affect in cis the chromatin conformation of the region, impairing the normal interactions between the enhancers and gene promoters, resulting in lower CDKN1C expression.
Recommendations for molecular testing
Most patients with BWS and SRS carry epigenetic defects; in these cases, the risk of recurrence is low. However, distinguishing between ‘isolated’ epigenetic defects and genetic mechanisms is of particular importance because of its impact on genetic counselling. Most diagnostic laboratories use Methyl Sensible-MLPA, which can directly identify both CNVs and LOM/GOM, to test for BWS and SRS. However, other methylation-based techniques are also used, which only indirectly detect CNVs (because of the impact of the duplications/deletions on the methylation indices). In these cases, an additional technique must be used to detect such CNVs.43 44
The utility of microarray analysis, in particular SNP-arrays, in patients with well-characterised BWS and SRS has been highlighted by several studies, leading to the identification of new molecular defects.28 45 Furthermore, SNP-arrays can be helpful in clarifying the molecular diagnosis in patients with BWS, especially to discriminate between pUPD and duplications.45
In addition to SNP-arrays, chromosomal and subsequent FISH analysis are required for the characterisation of duplications of more than 300 kb, as neither SNP-arrays, nor MLPA can detect balanced rearrangements or locate them on a chromosomal segment (as illustrated by patient 4, for whom karyotype and FISH analysis revealed a nonreciprocal balanced translocation in the healthy mother). This characterisation is essential for providing appropriate genetic counselling to these families.
In conclusion, we report 17 new duplications of the 11p15 region (spanning either both domains or all or part of only one domain) with their associated methylation profiles and phenotypes. These duplications are rare in both BWS and SRS. Nevertheless, the description of these rare molecular anomalies is of particular importance for describing the putative functional consequences of the duplications to explain the associated phenotypes and provide accurate information for genetic counselling.
Acknowledgments
We would like to thank the patients and their families; Jennifer Salem; the RSS/SGA Research & Education Fund and the Magic Foundation (Dayna Carney, www.magicfoundation.org) for their precious help to collect the clinical data for the US patients. We would like to thank Nathalie Thibaud, Cristina Das Neves, Marilyne Le Jule, Françoise Langlet, Fabienne Rajhonson, Corinne Mach, Valérie Olin, Sabrina Karagic, Elodie Lejeune, Aurélie Lafitte, Claude Estrade for their technical assistance for genetic analyses. We would also like to thank the Silver Russell Patients’ association from France (Association Française des Familles ayant un enfant atteint du Syndrome Silver-Russell ou néPetit pour l'Âge Gestationnel, AFIF SSR/PAG, www.silver-russell.fr). This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and Université Pierre et Marie Curie (UPMC-Paris 6). IN and YLB are members of the European Union’s Seventh Framework Programme FP7/ITN Ingenium 2007–2013.
References
Footnotes
Contributors SH collected the data and wrote the first draft. SC-B, J-PS and BK analysed and interpreted the cytogenetic analyses. MDH, SA, SR, CM, MLPL, BD, CH, RSN, PS, LVM, VT, EG and YLB recruited the patients from the different centres. FB and IN coordinated the study, recruited the patients in Trousseau hospital (Paris) and revised substantially the draft. All authors approved the final version and agreed to be accountable for all aspects of the work.
Competing interests None declared.
Patient consent Guardian consent obtained.
Ethics approval CCP Ile de France.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice Thsi article has been corrected since it was published Online First. The affiliations of RSN have been corrected.