Article Text

Abstract
Background Orodental diseases include several clinically and genetically heterogeneous disorders that can present in isolation or as part of a genetic syndrome. Due to the vast number of genes implicated in these disorders, establishing a molecular diagnosis can be challenging. We aimed to develop a targeted next-generation sequencing (NGS) assay to diagnose mutations and potentially identify novel genes mutated in this group of disorders.
Methods We designed an NGS gene panel that targets 585 known and candidate genes in orodental disease. We screened a cohort of 101 unrelated patients without a molecular diagnosis referred to the Reference Centre for Oro-Dental Manifestations of Rare Diseases, Strasbourg, France, for a variety of orodental disorders including isolated and syndromic amelogenesis imperfecta (AI), isolated and syndromic selective tooth agenesis (STHAG), isolated and syndromic dentinogenesis imperfecta, isolated dentin dysplasia, otodental dysplasia and primary failure of tooth eruption.
Results We discovered 21 novel pathogenic variants and identified the causative mutation in 39 unrelated patients in known genes (overall diagnostic rate: 39%). Among the largest subcohorts of patients with isolated AI (50 unrelated patients) and isolated STHAG (21 unrelated patients), we had a definitive diagnosis in 14 (27%) and 15 cases (71%), respectively. Surprisingly, COL17A1 mutations accounted for the majority of autosomal-dominant AI cases.
Conclusions We have developed a novel targeted NGS assay for the efficient molecular diagnosis of a wide variety of orodental diseases. Furthermore, our panel will contribute to better understanding the contribution of these genes to orodental disease.
Trial registration numbers NCT01746121 and NCT02397824.
- Genetics
- Diagnostics tests
- Molecular genetics
- Genetic screening/counselling
- Genome-wide
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Orodental disorders encompass a number and variety of diseases that affect the teeth and oral cavity. Broadly, these disorders can be classified into anomalies of tooth number, shape and size (eg, hypo/oligo/ano-dontia (collectively selective tooth agenesis (STHAG)), microdontia, globodontia), anomalies of tooth structure (eg, amelogenesis imperfecta (AI), hereditary dentin disorders) and anomalies of tooth eruption. The prevalence of these disorders varies from relatively common (4.2% for hypodontia in the Caucasian population)1 to extremely rare (1 in 100 000 for dentin dysplasia (DD) type 1).2
Orodental disorders can have a genetic, environmental or multifactorial basis.3 ,4 Although evidence demonstrates a role for environmental pollutants such as dioxins and fluoride in developmental enamel defects,5 ,6 a number of studies have also demonstrated a strong genetic aetiology for several orodental diseases (reviewed in refs. 7–9). Among the >5000 known genetic syndromes, >900 have orodental/craniofacial features.10 Even in the case of isolated orodental diseases, significant genetic heterogeneity exists, with several of the same genes being involved in isolated and syndromic forms of disease. For instance, mutations in eight genes have been implicated in STHAG (PAX9, MSX1, LTBP3, AXIN2, WNT10A, EDA, EDARADD and EDAR),11–17 of which several (MSX1, WNT10A, EDA, EDARADD and EDAR) have also been linked with forms of ectodermal dysplasia.18–20 Similarly, mutations in a number of genes have been implicated thus far in AI, of which 10 cause an exclusively dental phenotype (ENAM, WDR72, KLK4, AMELX, MMP20, FAM83H, AMBN, ITGB6, SLC24A4 and c4orf26),21–30 some cause syndromic disease with AI (LTBP3, FAM20A, CNNM4, ROGDI, STIM1 and FAM20C),31–36 and yet others account for isolated and syndromic AI (COL17A1, LAMA3, LAMB3 and DLX3).37–40 Indeed, the pattern of inheritance and penetrance associated with each gene also varies. This wide range of heterogeneity can render genetic diagnosis challenging.
Yet, the early molecular diagnosis of orodental disorders is important as it can improve patient care. For instance, mutations in AXIN2 that cause STHAG have been shown to predispose carriers to colorectal cancer.12 Early diagnosis of AXIN2 mutations can hence alert clinicians to counsel patients to have regular colonoscopies. Similarly, the identification of mutations in FAM20A in patients presenting with AI can prompt a renal investigation for the management of nephrocalcinosis.41
Targeted next-generation sequencing (NGS) has proven extremely beneficial clinically for the molecular diagnosis of a number of genetically heterogeneous disorders, such as hearing loss, mitochondrial disease, intellectual disability (ID), neuromuscular disorders and Bardet–Biedl syndrome.42–46 Better coverage, lower cost and relative ease of data interpretation have made it more commonplace for routine clinical use than whole-exome sequencing (WES) and whole-genome sequencing (WGS).47
We have developed the first targeted NGS panel for the molecular diagnosis of isolated and syndromic orodental disorders. We demonstrate the utility of this panel in the molecular diagnosis of a variety of orodental disorders. In a cohort of 103 patients (101 unrelated) without a known molecular diagnosis referred to the Reference Centre for Rare Diseases with Oro-dental Manifestations, we were able to provide a definitive molecular diagnosis in 39% of patients in known disease-causing genes. The identification of mutations in genes underlying syndromic forms of orodental disease highlights the potential benefits of a complete oral investigation in the diagnosis of rare genetic diseases. The aim of this article is to demonstrate the utility of this targeted NGS assay for the diagnosis of mutations in known orodental disease genes. However, the potential for the discovery of novel genes is addressed in the discussion.
Methods
Patients
Patients were referred to the Reference Centre for Rare Diseases with Oro-dental Manifestations (Strasbourg, France) by dentists, paediatricians and geneticists from 13 hospitals and 32 private practices in France, Germany and Morocco. The inclusion criterion for the study was the presence of an orodental anomaly, defined as an anomaly of the mouth, including teeth and surrounding structures such as the periodontium (alveolar bone, ligament and gingivae), as well as defects of lip and palate formation. Patients with known mutations used for the validation assay were previously diagnosed either in clinical (Laboratory for Genetic Diagnosis, Strasbourg University Hospital) or research laboratories. DNA was obtained from peripheral blood or saliva samples (Oragene DNA, DNA Genotek, Canada). Patient phenotype was recorded using D[4]/Phenodent (http://www.phenodent.org).
Gene selection and targeted capture design
Genes were selected based on their involvement in human diseases with orodental phenotypes, mutation in animal models presenting orodental disorders,48 ,49 expression in the developing mouse tooth50 and known role in tooth development. Two versions of the gene panel were developed, v1.0 and v2.0. v1.0 was used for patients V1.01–V1.16, whereas v2.0 was used for patients V2.01–V2.95. Complementary RNA capture probes were designed against all coding exons and 25 bp of flanking intronic sequence in order to cover splice junctions of these genes using the SureDesign portal (https://erray.chem.agilent.com/suredesign, Agilent, USA).
Library preparation, sequencing and data analysis
Targeted regions were captured using a Custom SureSelectXT2 in-solution target enrichment kit (Agilent) and libraries were prepared for sequencing (2×100 bp) on the HiSeq2500 (Illumina, USA) following the manufacturer’s instructions. For v1.0, 16 samples were multiplexed per lane for sequencing, whereas for v2.0, 32 samples were multiplexed per flow-cell lane. Read alignment, and variant calling and annotation were performed using standard methods. Briefly, reads were aligned to the GRCh37 reference genome using Burrow–Wheeler aligner (v0.7.5a)51 ensuring tagging of multi-mapped reads, and duplicates were marked with Picardv1.102 (http://picard.sourceforge.net). Indel realignment, base quality score recalibration and variant calling were performed with the GATK Toolkit v3.1 using hard-filtering parameters.52–54 Variants were annotated using snpEffv.3.4.55 Variant frequencies were compared with an internal exome database and prioritised using VARank.56 Variants were prioritised by allele frequency (<1% in the Single Nucleotide Polymorphism database (dbSNP137), 1000 Genomes database,57 Exome Variant Server (EVS) database58 and our internal database, except for non-syndromic STHAG, for which we used a cut-off of <4%) and predicted functional effect (frameshift, invariant splice sites, non-synonymous and splice affecting mutations). Missense mutations were evaluated for pathogenicity bioinformatically using Sorting Intolerant from Tolerant (SIFT), PolyPhen, MutationTaster and amino acid conservation.59–61 Splice affecting mutations were evaluated bioinformatically using Human Splice Finder, MaxEntScan, NNSplice, Gene Splicer, SSF, Rescue ESE and ESE Finder.62–67 CNVs were detected as previously described.44 ,45 Variant pathogenicity was interpreted according to the American College of Medical Genetics guidelines.68 Evidence used to establish pathogenicity is provided in online supplementary figures S1–S45.
Mutation validation
All mutations were validated by Sanger sequencing (GATC, Germany). Segregation analyses were performed whenever DNA was available for additional family members. Single-molecule PCR followed by Sanger sequencing was used to phase biallelic mutations when parental DNA was unavailable.69 ,70 Deletions were validated by qPCR. The region of interest and an internal control region (RPPH1) were amplified from 2 ng of genomic DNA from the patient and a control using the iQ SYBR Green Supermix (170-8880, Biorad, USA) on a CFX96 Real-Time System (Biorad). Data were analysed using the Pfaffl method.71
Results
Validation of v1.0 of the NGS panel
As a proof of principle, we created a primary version (v1.0) of a custom NGS panel that targets 560 known and candidate genes (2.36 Mb) in orodental disorders. Of these genes, probes for 175 known and strong candidate genes (0.81 Mb) were designed to have superior coverage (diagnosis subpanel), whereas the remaining candidate genes constituted a ‘Discovery subpanel’ (see online supplementary tables S1 and S2). We validated v1.0 in a cohort of 16 patients: patients V1.01–V1.08 with isolated and/or syndromic orodental diseases with known mutations as determined by previous candidate gene Sanger sequencing, WES, or array CGH (see online supplementary table S3) and patients V1.09–V1.16 without a known molecular diagnosis. Patients V1.01–V1.08 were selected to include a variety of heterozygous, homozygous and hemizygous mutations (substitutions, indels, a large heterozygous deletion) at eight different loci in order to test the sensitivity of the gene panel to different types of genetic alterations. Using our computational pipeline, 10 of 10 mutations, including a large heterozygous deletion on the X chromosome (see online supplementary table S3), were detected in a blinded manner by a bioinformatician who was unaware of the molecular alteration but was aware of the clinical diagnosis for each patient. The bioinformatician was also blinded to the mode of inheritance in order to simulate a diagnostic scenario where such information is often unavailable. Samples V1.09–V1.16 were selected to cover a variety of orodental phenotypes (AI, dentinogenesis imperfecta (DGI), DD, STHAG) to determine the potential to identify unknown mutations with our panel. The molecular results from samples V1.09–V1.16 are presented and discussed in tables 1–3, together with the results from v2.0 of the gene panel presented below. Briefly, pathogenic or likely pathogenic mutations were identified in six of eight samples among samples V1.09–V1.16 (tables 2 and 3).
On average, in this validation cohort, by multiplexing 16 samples per lane of a sequencing flow cell, 2.2 Gb of sequence was generated per sample, giving a mean coverage of 365×, with 95.31% nucleotides covered at ≥50× (see online supplementary table S4). The mean coverage of the diagnostic subpanel was 404×, with 98.94% of nucleotides covered at ≥20×, permitting confident diagnosis of mutations in these genes, for which we set a minimum coverage threshold of 20× (see online supplementary table S4). A small percentage of targeted regions (33.3 kb, 1.4%) had an average coverage <20× across samples due to high guanine-cytosine (GC) content or pseudogenes with highly similar sequences (see online supplementary table S5). The majority of these regions were in the discovery subpanel (28.1 kb). Under-representation of high GC content and pseudogenic regions is a known issue with probe-based targeting strategies. However, these gaps can be filled in by Sanger sequencing for regions considered to be relevant on a case-by-case basis. On average, we detected ∼2648 variants/sample, of which ∼87 variants/sample were rare (<1% in dbSNP137, 1000Genomes, EVS and an in-house database). Among these rare variants, we identified on average per sample ∼20 missense changes, 0.38 nonsense mutations, 0.25 splice-site changes and 0.56 frameshift-inducing indels (see online supplementary table S6).
Development of v2.0 of the NGS panel
Having validated v1.0 of our NGS gene panel, we included 25 additional genes to the discovery subpanel to create a second version of the panel (v2.0) that included 585 genes (2.47 Mb). These additional genes were included due to their implication in animal models of orodental disease that was unknown when designing v1.0 of the panel v2.0 of the panel was used to sequence 95 patients (V2.01–V2.95). Furthermore, since the average coverage achieved with v1.0 was more than sufficient for confident molecular diagnosis, with v2.0, 32 samples were multiplexed per well of a flow cell lane in order to reduce sequencing costs while ensuring a minimum average coverage of 100× per sample. Sequencing output with v2.0 is shown in online supplementary table S7. Briefly, we achieved a mean coverage of 179× overall, with 97.2% of the targeted region covered at ≥20×. Furthermore, the diagnostic panel had an average coverage of 211× with 97% of bases covered at ≥50×.
Screening a cohort of patients with diseases with orodental involvement
A description of the final cohort of 103 patients (101 unrelated) without a known mutation sequenced with v1.0 (V1.09–V1.16) and v2.0 (V2.01–2.95) and the diagnostic yield by disease category is shown in table 1. Isolated AI was the most common disorder in the cohort (50%), followed by isolated STHAG (20%). We also included several patients with syndromic forms of AI, ranging from well-defined syndromes, such as Enamel Renal Syndrome, to undefined and suspected syndromes, hoping that a molecular diagnosis may aid difficult clinical diagnosis. The remaining cases consisted of patients with isolated or syndromic dentin disorders, syndromic STHAG, suspected otodental dysplasia and primary failure of tooth eruption. Since AI and STHAG can be inherited in an autosomal-dominant (AD), autosomal-recessive (AR) or X-linked fashion, the mode of transmission was unclear for the majority of cases. Sixty-six cases showed sporadic disease, whereas 35 showed familial transmission. The mode of inheritance could be confidently inferred only in 18 cases, 15 as AD due to the presence of other affected family members, 2 as AR due to the presence of consanguinity between the parents of the index case and 1 as X-linked. Due to the large number of cases without a known mode of inheritance of disease, a common situation in diagnostic cohorts, variant filtration was performed for all samples for all modes of inheritance. Overall, we had a definitive molecular diagnosis in known genes in 39 cases (39%) and discovered 21 novel pathogenic variants. All pathogenic mutations identified in this cohort are listed in table 2. Likely pathogenic mutations and other variants are shown in table 3. The distribution of pathogenic variants identified with respect to the disease category and the number of cases corresponding to each mutated gene is shown in figure 1.
Cohort description and diagnostic yield per disease category
Pathogenic mutations identified in patients without a known mutation
Likely pathogenic variants and other variants identified in patients without a known mutation
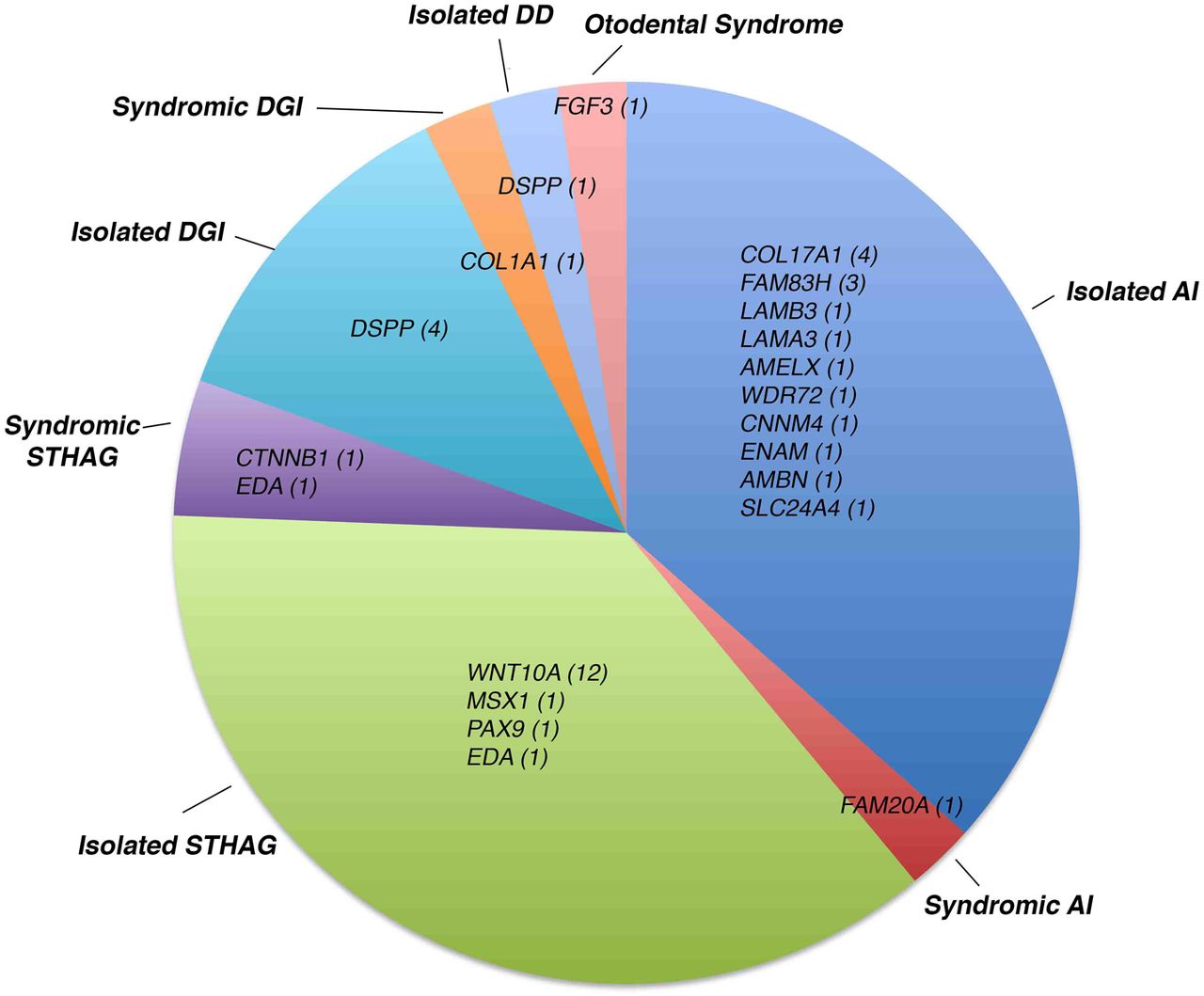
Distribution of pathogenic variants identified by disease category. The genes in which pathogenic mutations were identified are shown with respect to the disease in which they were identified. In parentheses next to each gene is the number of independent patients in whom mutations were identified in the gene. This figure includes results from patients V1.09–V1.16 and V2.01–V2.95. DD dentin dysplasia; DGI, dentinogenesis imperfecta; STHAG selective tooth agenesis.
Isolated AI: COL17A1 mutations are a frequent cause of AI
Of the 50 unrelated cases with confirmed isolated AI, we identified the causative mutation in 14 cases (27%). Surprisingly, the most frequently mutated gene in our cohort was COL17A1, with four independent patients segregating pathogenic mutations in this gene (of which three are novel). In the biallelic state, mutations in COL17A1, encoding a structural component of hemidesmosomes, cause junctional, non-Herlitz-type epidermolysis bullosa (EB), a skin blistering disorder with associated dental enamel defects.85 However, rare cases of isolated ADAI in heterozygous carriers have been reported.37 ,86 Similarly, heterozygous mutations in LAMA3 and LAMB3, which also encode hemidesmosomal components, can cause ADAI, whereas biallelic mutations in these genes cause EB.38 ,39 We found one family segregating a known heterozygous LAMB3 nonsense mutation. These patients showed a similar enamel phenotype characterised by a hypoplastic form of AI with thin enamel and a characteristic pitting of the enamel that varied in severity (figures 2A–H and 3C). Depending on the extent of pitting, the enamel surface was sometimes rough. Secondary extrinsic colouration of the pits rendered them more visible. Interestingly, patient V2.82 had two unlinked mutations, one maternally inherited novel COL17A1 splice mutation (c.1141+1G>A) and a novel LAMA3 frameshift-inducing deletion (p.I2159Mfs*46) that is absent in her mother (figure 2I). The father of the patient was unavailable for testing but was described as unaffected at the time of ascertainment. The c.1141+1G>A mutation is predicted to cause either in-frame skipping of exon 14 (which encodes a part of the intracellular domain) or retention of intron 14 that would subsequently introduce a premature termination codon (PTC) and likely nonsense-mediated decay (NMD) of the transcript. Patient V2.82's enamel phenotype is more severe than that of her mother, suggesting digenic inheritance in this individual. Patient V2.82 has hypoplastic AI with clearly visible pitted enamel in the vestibular, lingual and palatal surfaces of premolars and molars and a white colouration following Retzius striae visible in the permanent incisors (figure 2A, B). Her mother's dentition, in contrast, shows more discrete signs of AI and fewer pits are visible on the buccal surfaces of premolars (figure 2C, D). Digenic mutations in LAMB3 and COL17A1 that modify the severity of the EB phenotype have been previously reported.87 However, this is the first report of seemingly digenic inheritance in ADAI.
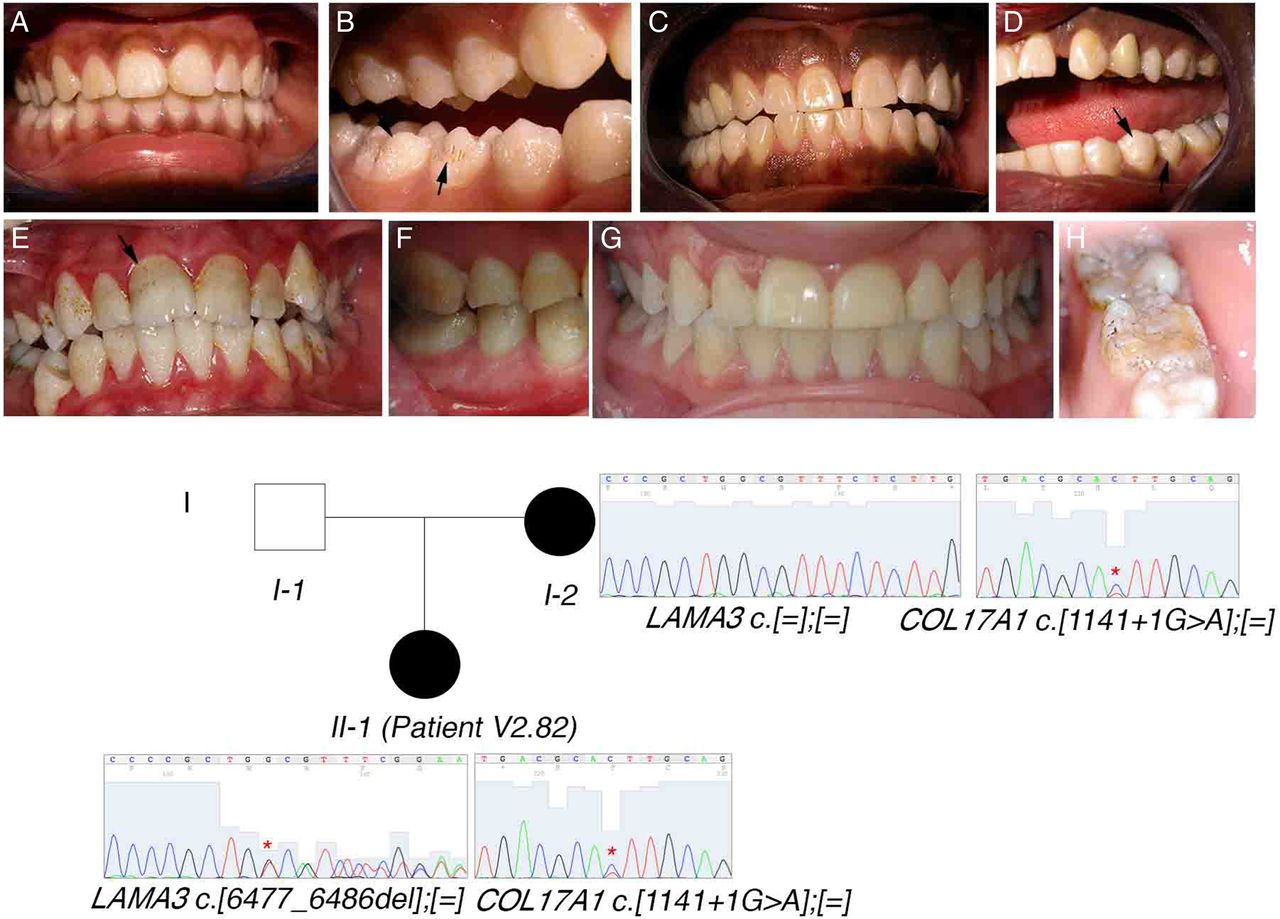
COL17A1 mutations show a characteristic enamel phenotype in autosomal-dominant amelogenesis imperfecta. (A–H) Photos of the enamel phenotypes of individuals with COL17A1 mutations. (A, B) Patient V2.82. (C, D) Mother of patient V2.82. (E, F) Patient V2.09. (G, H) Patient V2.48. Arrows mark pits in the enamel that are characteristic of COL17A1 mutations. Extrinsic colouration makes these pits more visible in (B), (E) and (H). (I) Seemingly digenic inheritance of amelogenesis imperfecta in patient V2.82. Asterisks on the sequence chromatograms mark the mutated nucleotides.

{kind=link}
{kind=link}
{kind=link}
The variety of isolated amelogenesis imperfecta phenotypes seen in this cohort. Representative images of the enamel phenotype associated with mutations in different genes. The mutated gene and patient number are indicated in each panel.
We also identified a family with a novel homozygous splice-site mutation (c.532–1G>C) in AMBN. To date, only a single family has been reported to carry mutations in this gene.27 Similar to the patients in the initial study, our patient too exhibited hypoplastic AI with very limited enamel as seen in the unrestored yellow-tinged premolars and permanent molars. However, the tooth surfaces did not seem as pitted as described previously (figure 3F).27 This mutation is predicted to alter mRNA splicing by inducing either the skipping of exon 7 or the retention of intron 6. Exon skipping would lead to in-frame deletion of exon 7, which encodes a domain involved in heparin and fibronectin interaction that is thought to be important for the interaction of AMBN with dental epithelial cells.88 ,89 Intron retention would introduce a PTC that is likely to cause NMD of the mRNA. Additionally, a cryptic acceptor splice-site is predicted bioinformatically to be activated in exon 7 that would cause partial out-of-frame skipping of exon 7 and a subsequent PTC (see online supplementary figure S11). Thus, this mutation is predicted to cause at least a partial loss of AMBN function. We also identified novel mutations in WDR72, AMELX and ENAM, and a known deletion of the last three exons of SLC24A4 (table 1). Representative images of the enamel phenotype associated with mutations in each gene are shown in figure 3. Similar to previous reports, WDR72 and SLC24A4 mutations caused hypomineralised, hypomature AI with brownish/yellowish discoloured teeth.22 ,29 The enamel showed a lack of contrast with the underlying dentin upon X-ray imaging (data not shown). The softer enamel seen in the patient with WDR72 mutations was subject to wear, whereas the enamel in the patient with the SLC24A4 mutation was opaque. Patients with AMELX, LAMB3 and ENAM mutations had hypoplastic AI with yellow discolouration and smaller teeth due to thinner enamel.
Interestingly, we identified a novel homozygous missense mutation (p.V499M) in CNNM4 in patient V2.05 who was born to first-cousins and was referred for isolated AI. This mutation affects a highly conserved residue in a functional cystathionine beta-synthase domain of the protein and is predicted bioinformatically by three algorithms (SIFT, PolyPhen and MutationTaster) to be deleterious (see online supplementary figure S2). However, mutations in this gene are known to cause Jalili syndrome, which is characterised by a combination of AI and cone-rod dystrophy.33 ,90 A subsequent full-field electroretinography revealed a marked loss of cone response and a less severe loss of rod response in the patient, thus confirming the Jalili phenotype (see online supplementary figure S2E, F).
Syndromic AI
We confirmed the molecular diagnosis in one case of Enamel Renal Syndrome (patient V2.06). This patient was homozygous for a novel frameshift-inducing deletion in FAM20A (c.1106_1107del).
In patient V2.15, referred for the management of AI in the context of suspected spondyloepiphyseal dysplasia (SED), we identified a known pathogenic missense mutation (p.T312S) and a likely pathogenic intronic variant (c.121-31) in GALNS (mutated in mucopolysaccharidosis (MPS) IVA) predicted to create an intronic splice enhancer (see online supplementary figure S42). However, the biological effect of this intronic mutation warrants further investigation. No deleterious mutations were identified in CHST3 or COL2A1 and a differential diagnosis of mucolipidosis type III was eliminated due to normal hexosaminidase, alpha-l-fucosidase and beta-d-glucuronidase levels (data not shown). Similar clinical presentation of SED and MPS IVA has been previously demonstrated;91 therefore, our results suggest a potential differential diagnosis of MPS IVA in this patient. In the case of patient V2.49, referred for the management of AI linked to MPS IVA,92 ,93 we identified only a single de novo known pathogenic mutation in GALNS (p.R386C), suggesting the presence of a second mutation in intronic or regulatory regions, or a structural mutation missed by our pipeline. The presence of an undetected second mutation is consistent with the virtually absent GALNS activity in the patient's leucocytes (1 nmol/h/mg protein) (data not shown). Both these patients exhibited hypoplastic AI with thin striated and pitted enamel and subsequently flattened buccal surfaces (see online supplementary figure S45) consistent with previous reports of the enamel phenotype seen in MPS IVA,94 further supporting a diagnosis of MPS IVA in both patients. However, additional tests are necessary to confirm this diagnosis.
Isolated and syndromic STHAG
We identified the causative mutation in 15/21 (71%) cases of isolated STHAG. The majority carried mutations in WNT10A either in a heterozygous, homozygous or compound heterozygous state as has been previously reported.77 ,79 Fortuitously, we were also able to provide a molecular diagnosis for a patient presenting with ID who was referred for the management of hypodontia. The patient (V2.87) had agenesis of five incisors, two maxillary incisors and three mandibular permanent incisors (agenesis 12, 22, 41, 31 and 32) and had no family history of hypodontia or ID. We identified a novel heterozygous nonsense mutation in CTNNB1 (p.Tyr333*), which encodes beta-catenin, a member of the WNT signalling pathway, which is essential during multiple stages of tooth development.95 Recent reports have established a role for heterozygous CTNNB1 mutations in ID characterised by mild to severe ID, autism spectrum disorder childhood hypotonia with progressive spastic diplegia, microcephaly and significant additional craniofacial and brain abnormalities, which is compatible with this patient's phenotype (data not shown).96 ,97 However, the orodental phenotype in this syndrome has not been studied in detail. Our report suggests that patients with ID due to CTNNB1 mutations may require a dental examination and management of hypodontia. Furthermore, this case highlights the benefit of an interdisciplinary approach to patient care and the potential utility of a full dental examination in syndromes with craniofacial involvement.
Dentinogenesis imperfecta and dentin dysplasia
Among the six independent cases of isolated dentin disorders, we identified the causative mutation in five. All patients carried mutations in DSPP, the only gene implicated thus far in isolated DD/DGI.2 The majority of mutations were present in the repeat-rich exon 5 that is often refractory to Sanger sequencing and hence until now excluded from routine diagnostic sequencing.98 We achieved an average coverage of 771× and 408× over exon 5 of DSPP with v1.0 and v2.0, respectively. Hence, our assay provides an effective means to screen for mutations in the last exon of DSPP. Our bioinformatic pipeline detected the majority of mutations; however, one mutation (c.3682_3686del) was missed due to the difficulty of calling variants in structurally complex regions (personal communication, MKP, Broad Institute GATK Team), but was detected upon visual inspection of the reads. We also identified a novel frameshift-inducing deletion in COL1A1 in a family segregating AD osteogenesis imperfecta with DGI.
Otodental dysplasia
We identified a heterozygous deletion of the first exon of FGF3 in a family segregating suspected AD otodental dysplasia, which is characterised by globodontia and hearing loss, sometimes segregating with colobomas, most likely due to the deletion of the adjacent FADD gene.99 This deletion was validated by qPCR on genomic DNA, which showed the presence of the deletion in patient V2.03 as well as in his affected father (see online supplementary figure S38).
Discussion
We have developed a targeted NGS assay for the diagnosis and discovery of mutations underlying rare genetic disorders with orodental manifestations. We achieved high sequencing coverage in the targeted regions, 179× average coverage with 97.2% of the targeted region covered at ≥20× with V.2.0. In a cohort of 101 unrelated patients with a variety of orodental genetic disorders of unknown genetic aetiology, we were able to detect the underlying pathogenic mutation in 39 cases (39%) in known genes.
Targeted NGS gene panels are being widely used for the diagnosis of a variety of genetic disorders, including ID, mitochondrial disorders and neuromuscular diseases.42–44 However, to the best of our knowledge, this is the first report of a targeted NGS gene panel for orodental disorders. Therefore, this diagnostic tool responds to a veritable need within the dental genetics community. Furthermore, two concrete examples from our cohort suggest the utility of this tool to the wider medical genetics community. First, we were able to diagnose a mutation in CTNNB1 in a patient with ID and tooth agenesis, suggesting the potential utility of our assay in screening for mutations in ID cases with orodental involvement. Second, the detection of a CNNM4 mutation in a patient referred for isolated AI led to an ophthalmological investigation for cone-rod dystrophy and the confirmation of Jalili syndrome. These cases highlight how the oral consultation can be a port of entry for the diagnosis and management of patients with rare genetic diseases, especially in light of the involvement of the same genes in syndromic and isolated forms of orodental disease and the sometimes non-evident extra-oral signs associated with some diseases.
Our targeted NGS panel targets fewer genes than WES/WGS and may thus exclude potential disease-causing genes. Yet, it has several advantages in a clinical setting. First, it provides higher sequencing coverage in the targeted regions than WES/WGS. A comparison of the coverage of the same regions achieved with v2.0 of our gene panel and a commercially available WES kit demonstrates that the coverage from WES falls short of the requirements for confident molecular diagnosis, that is, only 89.09% of targets covered at ≥20× with WES vs 97.23% of targets covered at ≥20× with our panel (see online supplementary figure S46 and table S8). Second, due to the smaller number of variants identified by targeted sequencing (∼2600 variants with our panel vs ∼30 000 with WES), variant interpretation is relatively simpler. This is also true for CNVs—we were able to detect and validate a single exon deletion in FGF3. Finally, the cost, amount of time for data analysis and interpretation, and data storage requirements are lower with targeted NGS than with WES/WGS approaches, making it an attractive alternative for clinical implementation. Alternatively, our assay could serve as a primary mutation-screening tool to exclude mutations in known genes before performing WES/WGS.
We expected that the inclusion of candidate genes selected based on their expression in developing mouse teeth50 and their implication in animal models of orodental disorders would permit the identification of novel genes mutated in these disorders. In order to identify potentially novel genes in patients lacking mutations in known genes, we performed a preliminary analysis focusing on rare loss-of-function variants (nonsense, invariant splice site and frameshift variants) that were present in the same gene in at least two independent families with the same phenotype. Such a strategy was previously used successfully to identify a novel gene, SETD5, in ID.100 However, this preliminary analysis did not yield any positive findings. Indeed, this analysis was limited in that it did not consider missense mutations whose effect is less clear and was complicated by the lack of information regarding the mode of inheritance in the majority of cases. Also, in this analysis patients were regrouped by disease category (all mutation-negative AI patients, all mutation-negative STHAG patients) without subclassification based on fine phenotype (eg, hypoplastic vs hypomineralised AI and severity of STHAG), which may be important to include in order to overcome the difficulties posed by genetic heterogeneity.101 Furthermore, the rarity of mutations in some genes may also necessitate much larger cohorts. Indeed, future analyses of this cohort will try to address these limitations.
Among the 50 unrelated patients with confirmed isolated AI in our cohort, we had a molecular diagnosis rate of 27%. This is slightly lower than the diagnostic rates reported by other studies that performed candidate gene sequencing in large AI cohorts. By performing Sanger sequencing of six genes (FAM83H, ENAM, AMELX, MMP20, KLK4 and WDR72) in 71 families, Wright et al75 identified mutations in 26 families (37%). By sequencing the same six genes in an independent cohort of 39 AI kindreds, Chan et al102 identified mutations in 19 kindreds (49%). In the latter study, the diagnostic rate was shown to be dependent on the mode of transmission: X-linked>AD>AR>simplex cases. In our study, the majority of cases were simplex cases (31 cases), which may explain to a degree our lower diagnostic rate despite the inclusion of a larger number of known AI genes. Furthermore, the 50 patients selected for this study belong to a larger AI cohort of 71 patients, a part of which (including patients in this study) had been previously sequenced by candidate gene Sanger sequencing and in which mutations were identified in 11 patients103 (online supplementary table S3 and unpublished data). Therefore, the overall diagnostic rate in the 71-patient cohort is 35% (25 patients), which is closer to published diagnostic rates. In further contrast to the above-mentioned studies where FAM83H was the most frequently mutated gene in ADAI, COL17A1 was the most frequently mutated gene in ADAI in our cohort, with 8% of our AI cohort carrying deleterious mutations in COL17A1. Additionally, we demonstrate that the presence of unlinked mutations in COL17A1 and LAMA3 can modify the severity of AI, suggesting a digenic mode of AI inheritance associated with mutations in the hemidesmosomal components. Therefore, our findings are novel in demonstrating the frequency of COL17A1 mutations in ADAI. Finally, the large number of AI patients without mutations in known genes suggests that the genetic and allelic heterogeneity underlying AI is yet to be fully unravelled.
The diagnostic rate in syndromic AI was low in our cohort (7%). This is likely because of the inclusion of patients without a clear clinical diagnosis. These cases likely represent novel phenotypes with mutations in novel genes. Thus, WES may be better suited for such cases. Yet, the identification of a single de novo pathogenic mutation in GALNS in patient V2.49 can inform genetic counselling in this family. Similarly, the identification of one pathogenic and one potentially pathogenic mutation in GALNS in patient V2.15 permits an evidence-based investigation of MPS IVA by assaying leucocyte GALNS activity.104 Therefore, although sequencing did not provide a definitive molecular diagnosis in these cases, the results of this assay can contribute to orienting clinical investigations/counselling.
Among the 21 cases of isolated STHAG, we identified the causative mutation in 15 (71%). This diagnostic rate is comparable to that reported in the literature. By screening WNT10A, MSX1, PAX9, IRF6 and AXIN2 by Sanger sequencing in 34 patients with isolated hypodontia, van den Boogaard et al77 were able to identify the underlying mutation in 71% of cases. Similar to other reports, WNT10A accounted for the majority of mutations in our cohort (52%) and were also associated with ectodermal dysplasia (patient V2.54), with F228I, similar to other reports, being the most commonly mutated allele in our cohort, consistent with its population frequency (2.4% in European-Americans) and the prevalence of STHAG in this population (4.2%).77–79 ,105 ,106
In conclusion, we have developed the first targeted NGS gene panel for the diagnosis and discovery of mutations in genetic disorders with orodental involvement. This panel can be reliably used for the molecular diagnosis in known genes of a variety of genetic disorders and can serve as a primary screening tool before the application of WES/WGS. Additionally, this panel also provides the potential for the discovery of novel genes mutated in orodental disorders.
Acknowledgments
The authors would like to thank all patients and their families for participating in this study. They also thank Ms Vanessa Stoehr for administrative help, Dr Mathilde Huckert and Jorel Salomon for technical assistance, Drs Elise Schaefer and Sophie Scheidecker for help with obtaining clinical data, and Dr Yaumara Perdomo-Trujillo for the electroretinography examination. They would like to thank Drs Alain Verloes, Jean-Yves Sire, Jean-Pierre Clavert, Béatrice Walter, Patrick Lutz, André Nirrengarten, Isabelle Rian, Thibault Sibert, Laurence Lustig-Grimm, Daniel Jasiak, James Lespinasse, Jacques Hassid, Smail Hadj-Rabia, Gabriel Dominici, Laurent Riguet, Dan Lipsker, Jean Luc Alessandri, Florence Rousselet, Philippe Schoenlaub, François Serres, Etienne Mornet, Thomas Edouard, Bertrand Baumann, Klaus Dietrich, Yves Bolender, Sandra Mercier, Elisabeth Steichen-Gersdorf, Jean-Gabriel Chillès, Hadrien Bonomi Dunoyer, Philippe Jonveaux, Supawich Morkumed, Patimaporn Pungchanchaikul, Sameh Mabrouk, Noora Zouari, Ilhame Mamouni, Alicia Rossa, Caroline Delfosse, Sylvie Goldmann, Esther Levy, Pierre Wahl, Jean-Claude Schoeffler, François Charton, Pascale Meng, Pierre-Emile Spaeth, Marie-Claude Addor, Rene Serfaty, Anthony Kurtz, Renaud Rinkenbach, Alain Duret, Jehan Figier, Denys Chaigne, Marc Hildwein, Florence Jouanet, Olivier Grison, Marie-Noelle Duffard, Gautier Jouvenet Gilquin, Francois Undreiner, Gabrielle Baver, Isabelle Zumsteeg, Cloé Vorpahl, Alain Burtscher, Michel Kretz, Cyrielle Marc, Jean-Pierre Strauss, Elise Pilavyan, Pierre Bonnaure, and Celine Benarroch-Leininger, Rémy Mathis, William Bacon, Marie-Rose Javier, Bilal Ahmed and Elia Sfeir for referring patients. Sequencing was performed by the IGBMC Microarray and Sequencing platform, a member of the ‘France Génomique’ consortium (ANR-10-INBS-0009).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
Footnotes
Contributors Study concept and design: MKP, HD and AB-Z. Patient inclusion: MS, BL, DD, AD, AL, BG, MM-B, SLC, MM, SJ, FO, VV, J-LD, TD-B, A-SK, UM, BR, J-JM, J-PD, SO, IB-F, MMR, LM, AT, CJ, FG, J-CD, AC, MEA, SL, SS, NG, AD, BD, SF, EG, BF, MDM, YA, CT, FC, AB, MCM, HD and AB-Z. Data generation, analysis and interpretation: MKP, VG, SV, BJ, MD, SLG, VL-H, BG, MP, CS and AB-Z. Drafting of the manuscript: MKP and AB-Z. Critical reading of the manuscript: UM, AB and HD.
Funding This work was supported by grants from the French Ministry of Health (National Program for Clinical Research, PHRC 2008 N°4266 Amelogenesis imperfecta), the University Hospital of Strasbourg (API, 2009–2012, ‘Development of the oral cavity: from gene to clinical phenotype in Human’) and the EU-funded project (ERDF) A27 ‘Oro-dental manifestations of rare diseases,’ supported by the RMT-TMO Offensive Sciences initiative, INTERREG IV Upper Rhine programme. http://www.genosmile.eu
Competing interests None declared.
Patient consent Obtained.
Ethics approval French Ministry of Higher Education and Research (MESR).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All PCR primers and protocols are available upon request from the corresponding author.