Article Text

Abstract
Introduction Norrie disease is a rare X-linked congenital retinal vasculopathy that may be accompanied by sensorineural deafness, mental retardation, and other neurological deficits. Here we present a family in which Norrie disease co-segregated with either early-onset idiopathic pulmonary hypertension or sudden death preceded by a period of progressive dyspnea. Neither Norrie disease, nor its atypical variants described to date, have been associated with this extended clinical phenotype.
Methods and Results Molecular analysis of the Norrie disease gene (NDP) and adjacent loci was performed by multiplex ligation-dependent probe amplification and comparative genomic hybridisation. Affected males in this family showed an inherited hemizygous deletion restricted to NDP and two immediately telomeric genes, monoamine oxidase-B (MAO-B) and monoamine oxidase-A (MAO-A), which encode closely related enzymes that metabolize biogenic amines including serotonin, dopamine, and norepinephrine. Sequencing of the deletion junction showed an unusual pattern in which a region of microhomology flanked intervening genomic sequence.
Conclusion Because abnormalities of biogenic amines, particularly serotonin, have been implicated in the pathophysiology of pulmonary hypertension, we propose that presumed MAO deficiency in these patients may represent a novel risk factor for pulmonary hypertension, particularly forms with very early onset. Fine-mapping of other microdeletions at this locus may provide insights into additional mechanisms for nonrecurrent genomic rearrangements at this and other chromosomal loci.
- Norrie disease
- idiopathic pulmonary hypertension
- genomic rearrangement
- monoamine oxidase
- contiguous gene deletion syndrome
- clinical genetics
- molecular genetics
- neurology
- pulmonary hypertension
Statistics from Altmetric.com
- Norrie disease
- idiopathic pulmonary hypertension
- genomic rearrangement
- monoamine oxidase
- contiguous gene deletion syndrome
- clinical genetics
- molecular genetics
- neurology
- pulmonary hypertension
Introduction
Norrie disease (ND, OMIM 310600) is a rare neurodevelopmental disorder characterised by congenital blindness, progressive sensorineural hearing loss and cognitive impairment.1 It is caused by inactivating mutations in the gene encoding norrin (NDP), a ligand for the heterodimer of frizzled-4 (FZD4) and low-density lipoprotein receptor protein-5 (LRP5). In their activated state, these proteins are known to promote degradation of β-catenin in a tissue-specific pattern.2 Mutations in any of these three proteins produce a pattern of defective retinal vascularisation known as exudative vitreoretinopathy, characterised by persistently dilated retinal vessels and delayed regression of the retrolental hyaloid vessels. Studies of the NDP knockout mouse also reveal abnormally dilated vessels of the stria vascularis of the inner ear, a pathology that may mediate hearing loss in human subjects.3
NDP maps to the short arm of chromosome X (p11.4-p11.3) in tandem with the genes encoding monoamine oxidase A (MAO-A) and monoamine oxidase B (MAO-B). These enzymes metabolise intraneuronal and endothelial pools of biogenic amines including serotonin, dopamine and norepinephrine. The chromosomal region encompassing NDP, the MAO genes and other nearby genes including EFHC2 is prone to non-recurrent microdeletions.4–11 These genomic rearrangements are believed to give rise to ‘Norrie-plus’ clinical syndromes, or contiguous gene deletion syndromes, in which the typical features of Norrie disease co-segregate with other apparently unrelated features such as severe psychomotor retardation, microcephaly, growth retardation and/or seizures.7 9 12 13
Here we describe a family with a novel Norrie-plus syndrome in which idiopathic pulmonary hypertension co-segregated with ND. Molecular analysis revealed an inherited microdeletion encompassing NDP, MAO-A and MAO-B. Because abnormalities of biogenic amines, particularly serotonin, have been implicated in the pathophysiology of pulmonary hypertension, we propose that presumed monoamine oxidase deficiency in these patients may represent a novel risk factor for pulmonary hypertension, particularly forms with very early onset.
Clinical case
The proband (figure 1, II.4), fourth child of healthy, non-consanguineous parents, was born at 38 weeks' gestation with a birth weight of 7 lbs 6 oz. The pregnancy was complicated by uterine bleeding and abdominal pain, but otherwise the mother's medical history was unremarkable. At birth, the child was noted to have bilateral leucocoria, and further ophthalmological evaluation revealed an exudative vitreoretinopathy consistent with ND. Evaluation for cytomegalovirus (CMV) retinitis was negative. By age 3, he was noted to have marked developmental delay as well as somatic growth failure (height, weight and head circumference at less than the 10th centile): language was limited to a few words, and motor development was severely delayed (limited to sitting up with assistance). His clinical course was notable for seizures, controlled by carbamazepine, with EEG showing generalised spike wave abnormalities. Diffuse hypotonia and hypoactive reflexes were noted.
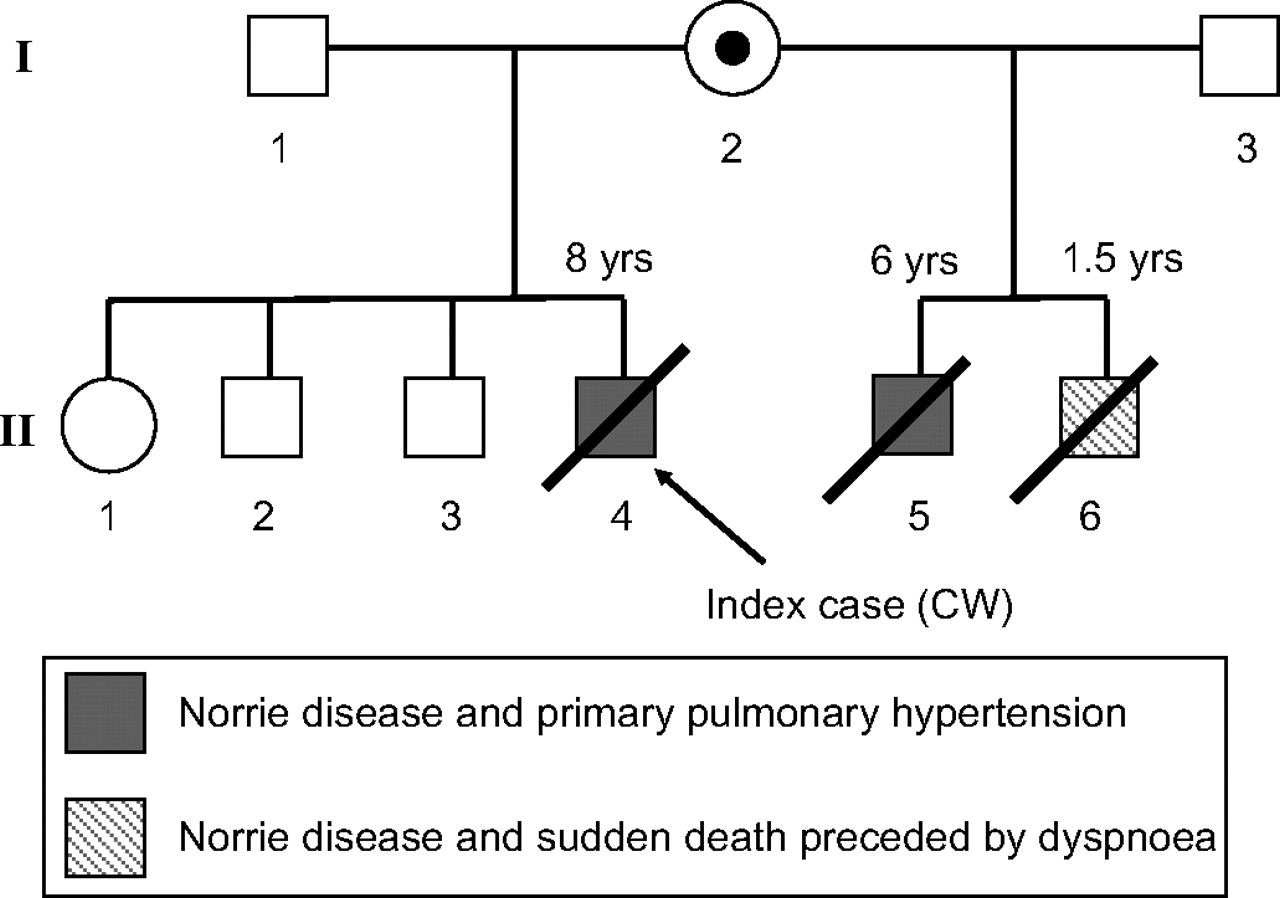
Pedigree of family showing co-segregation of Norrie disease and idiopathic pulmonary hypertension or sudden death. The mother (I.2) of the affected brothers (II.4, II.5 and II.6) is a carrier of the microdeletion shown in figure 2 and described in detail in the text.
At age 8, the boy was admitted for high-fever pneumonia accompanied by poor feeding and loose stools. The following day, his central nervous system status deteriorated, and, upon transfer to the paediatric intensive care unit, he was found to be markedly hypoxic with evidence of florid pulmonary hypertension. Transthoracic echocardiography revealed a markedly increased end-diastolic right ventricular anterior wall thickness (RVAWT) of 4.8 mm (normal age-specific range: 1.7–3.9 mm14) but no structural cardiac anomalies. The tricuspid valve was anatomically normal, but Doppler imaging measured a regurgitation velocity of 5.5 m/s, yielding an estimated pulmonary artery pressure of 130/100 mm Hg. There was no clinical evidence of systemic or portal hypertension, and CT imaging showed no evidence of a pulmonary embolus. The patient died 2 weeks later from progressive right-sided cardiac failure.
The proband's maternal half-brother (figure 1, II.5) had a remarkably similar clinical course. He was diagnosed with ND at age 2 and presented to the hospital with progressive dyspnoea and cardiac decompensation in the setting of pneumonia. Again, the clinical and imaging evidence favoured idiopathic pulmonary hypertension. Specifically, echocardiography showed a RVAWT of 4.2 mm and an estimated pulmonary artery pressure of 100/70 mm Hg. The patient showed a moderate but short-lived improvement in oxygenation status after trials of a prostacyclin analogue (epoprostenol) and a cGMP phosphodiesterase inhibitor (sildenafil). Both of these agents have been used with limited success in cases of pulmonary hypertension. The patient died at the age of 6 years.
A third brother (figure 1, II.6), diagnosed with ND at age 1, died suddenly at 18 months after a 3-day period of progressive dyspnoea. The family included three additional healthy siblings. The remainder of the family history, including that of the heterozygous carrier mother, was non-contributory.
Materials and methods
Multiplex ligation-dependent probe amplification (MLPA) and sequencing
Genomic DNA was extracted from peripheral blood specimens using standard manual methods (Qiagen, Valencia, California, USA). MLPA of NDP coding exons and flanking genomic DNA (probes: 2 Mb telomeric to exon 3 of NDP, 1 Mb telomeric to exon 3 of NDP, MAO-A, 100 kb centromeric to exon 1 of NDP, and EFHC2) were designed according to previously described methods.15 Further analysis of the BMPR2 gene for possible genomic deletions and rearrangements was performed by MLPA according to the manufacturer's instructions using the P093 Salsa MLPA HHT/PPH1 probe set (MRC-Holland, Amsterdam, The Netherlands). Sanger sequencing of the coding regions and exon–intron boundaries of BMPR2 were performed on a 3130xl Genetic Analyser (Applied Biosystems, Foster City, California, USA).
Comparative genomic hybridisation (CGH)
CGH was performed according to previously published methods of analysis using the Agilent 244K human genome oligonucleotide CGH microarray; all coordinates reflect human genome build 18 (G4411B; Agilent Technologies, Palo Alto, California, USA).
A high-resolution 1-million feature Agilent CGH microarray was designed for chromosome X. Probes with unique genomic mappings were used, where possible, to tile across chromosome X with a target mean of 100 bp between neighbouring probes. The custom probes were filtered by predicted melting temperature, unique genomic mappings, repeat content, and for the presence of known single-nucleotide polymorphisms. The DNA was fragmented, labelled and hybridised according to standard Agilent protocols. The array was scanned on an Agilent Scanner with 2 μm resolution. Intensity values were obtained using Agilent's Feature Extraction software and were subsequently analysed using Agilent's Genomic Workbench software to identify aberrant regions.
Results
Molecular analysis of NDP and adjacent loci was performed by MLPA after failed PCR amplification of the three NDP exons. DNA from two affected brothers available for testing (figure 1, II.4 and II.6) showed a hemizygous deletion restricted to the entire NDP, MAO-A and intervening MAO-B genes (figure 2A). Testing of the mother (figure 1, I.2) demonstrated that she was a carrier of the same deletion. Complete sequencing and MLPA analysis of the BMPR2 coding region, performed as part of the work-up for idiopathic pulmonary hypertension, revealed no pathogenic mutations.

{kind=link}
{kind=link}
(A) X-chromosome tiling microarray analysis of the proband (II.4) showing a hemizygous 494 kb loss spanning MAO-A, MAO-B and NDP at chromosome Xp11.3. The gene EFHC2 (X:44007129-44202923), reported in several other non-recurrent microdeletions at this locus, is ∼3 Mb centromeric to the centromeric end of the microdeletion found in the current case. (B) Sanger sequence analysis of the deletion junction showing regions of microhomology (underlined sequence) flanking intervening genomic sequence (X:43374051-43374058, human genome build 18). This pattern was present in all three family members available for testing (I.2, II.4 and II.6).
As with other reported microdeletions involving the Norrie locus,4–11 13 16–18 we could find no obvious segmental duplications that would appear to mediate genomic rearrangement through non-allelic homologous recombination (http://genome.ucsc.edu/index.html). To gain insight into other possible mechanisms that may have produced this microdeletion, we used an X-chromosome tiling microarray at 100 bp resolution. This analysis revealed a 494.1 kb deletion spanning X:43373198-43867296 (human genome build 18) in the proband (figure 2A). We used these coordinates to design PCR primers flanking the deletion and nested primers to allow sequencing across the deletion junction. Interestingly, we found a three-nucleotide region of microhomology flanking an eight-nucleotide AT-rich segment of intervening genomic DNA (figure 2B). This intervening segment was unlikely to be a sequencing artefact, as it was present in a second affected brother (figure 1, II.6) as well as the mother. Five normal controls failed to reveal an amplification product, as expected, and copy number variations spanning this region have not been reported (http://genome.ucsc.edu/index.html). Genome-wide array CGH of the proband at ∼10 kb resolution revealed no other pathogenic genomic variants (data not shown).
Discussion
Atypical variants of Norrie disease have been described as part of a possible contiguous gene syndrome (Norrie-plus syndromes) caused by microdeletions encompassing NDP and neighbouring genes at Xp11.3-11.4 including MAO-A, MAO-B and EFHC2. Although seizures, myoclonus, profound psychomotor retardation, stereotypies, microcephaly, growth retardation, delayed sexual maturation and cardiovascular abnormalities, particularly tyramine hypersensitivity, have been described in patients with Norrie-plus syndromes,5 7 9 11–13 16 18 including those with deletions similar to the one we found in our family, we are not aware of a case that includes idiopathic pulmonary arterial hypertension (IPAH) as part of the clinical picture.
IPAH is a rare proliferative disorder of the distal pulmonary arterioles leading almost invariably to death from irreversible right-sided heart failure or arrhythmia.19 The major criteria for clinical diagnosis include a mean pulmonary artery pressure >25 mm Hg at rest, or >30 mm Hg during exercise, in the absence of secondary factors such as cardiac anomalies, connective tissue disease, pulmonary embolus, cirrhosis or systemic hypertension. The underlying pathology may be unmasked clinically by a process that increases oxygen demand or compromises oxygen delivery such as a respiratory infection, as was apparently the case with two of the affected brothers in this family. The sudden death of the third affected brother (figure 1, II.6) may have been secondary to an arrhythmia that developed in the setting of clinically undetected right ventricular hypertrophy. Indeed, IPAH may be a cause of paediatric sudden death that is diagnosed only at autopsy.20 21 Unfortunately, autopsy material was not available from this family to confirm the diagnosis of IPAH histologically.
Of the two or three cases of IPAH diagnosed per million per year, 80–90% are apparently sporadic and the remainder are familial.19 Heterozygous inactivating mutations of the transforming growth factor-β-related receptor, BMPR2, constitute the largest known risk factor for both forms of IPAH. The later-onset forms of IPAH, typically seen in the setting of hereditary haemorrhagic telangiectasia, have been linked to inactivating mutations in ALK1 (activin receptor-like kinase-1) and ENG (endoglin), both of which can serve as coreceptors of BMPR2 in endothelial cells. The low penetrance of BMPR2 mutations (∼20%) and the minimal pulmonary pathology observed in unchallenged BMPR2 +/− mice22 23 suggest that additional genetic or environmental factors modify the onset or clinical course of IPAH.
A subset of patients (∼10%) who developed IPAH after exposure to fenfluramine or related compounds that increase serotonin signalling had predicted inactivating mutations in BMPR2 not found in normal controls,24 suggesting an interaction between the BMPR2 receptor and serotonin signalling. Further implicating the role of serotonin was the association between in utero exposure to selective serotonin reuptake inhibitors and persistent pulmonary hypertension of the newborn.25 The BMPR2 status of these patients was not studied. Consistent with the foregoing clinical observations, prolonged serotonin exposure was found to increase susceptibility to pulmonary hypertension in BMPR2-deficient mice, which otherwise had haemodynamic and histopathological features similar to wild-type mice under both normoxic and hypoxic conditions.22
The genes encoding MAO-A and MAO-B, isoenzymes that play an important role in the metabolism of biogenic amines such as dopamine, serotonin, epinephrine and norepinephrine, are positioned just telomeric to the NDP locus at Xp11.4. Several studies have reported microdeletions of variable extent in this chromosomal region, including the NDP, MAO-A and MAO-B genes. This pattern has been hypothesised to extend the clinical spectrum of Norrie disease to include features such as atonic seizures, autistic-like behaviour and severe psychomotor retardation.9 12 13 18 Biochemical evaluation of one such family showed a marked increase in plasma serotonin in the male proband and an intermediate increase in female carriers12; these results were presumed to reflect a dose-dependent effect of MAO deficiency on peripheral serotonin concentrations.8 12 26 Consistent with this prediction, the blood pressure of subjects with combined MAO-A and MAO-B deficiency showed markedly increased sensitivity to intravenous infusions of tyramine.12 27
Because of the previously described association between defective serotonin homoeostasis and IPAH, we hypothesise that deletion of the MAO genes in the three boys affected with Norrie disease predisposed them to IPAH, perhaps in the setting of a ‘second hit’ such as BMPR2 deficiency. Complete sequencing of the BMPR2 gene in the proband, however, revealed no pathogenic mutations, and we therefore presume that MAO deficiency, in concert with other uncharacterised genetic or environmental modifiers, produces the clinical picture of IPAH in this kindred. One or more of these interacting factors may reside in linkage disequilibrium with MAO-A, MAO-B or NDP, but, given that similar contiguous gene deletions have not been previously associated with IPAH, it appears more likely that the second hit assorted independently in this family.
To our knowledge, this is the first study to fine-map and sequence the junction of a microdeletion encompassing the NDP locus. Consistent with the absence of neighbouring repetitive sequence elements, a common substrate for genomic rearrangements mediated by non-allelic homologous recombination,28 our data suggest that the microdeletion in this family may have arisen from microhomology-mediated end joining, as has been recently described for fine-mapped copy number variants throughout the human genome.29 The unusual inclusion of a small stretch of intervening genomic DNA in the final deletion product suggests that a more complex recombination event may have occurred. Ongoing fine-mapping of other ND patient-associated microdeletions may provide more general insight into additional potential mechanisms for non-recurrent genomic rearrangements at this and other chromosomal loci.
Acknowledgments
We thank Dr Michael Powell of the Division of Child Neurology, Huntsville Hospital, Huntsville, AL, USA for providing a clinical description of the affected family, and the laboratory of James Loyd at the Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA for sequencing and MLPA analysis of the BMPR2 gene in the proband. We thank the laboratory of Dr Charles Lee (Brigham and Women's Hospital, Boston, MA, USA) for the design of the X-chromosome tiling microarray.
References
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.