Article Text

Abstract
Background SHROOM4 is thought to play an important role in cytoskeletal modification and development of the early nervous system. Previously, single-nucleotide variants (SNVs) or copy number variations (CNVs) in SHROOM4 have been associated with the neurodevelopmental disorder Stocco dos Santos syndrome, but not with congenital anomalies of the urinary tract and the visceral or the cardiovascular system.
Methods Here, exome sequencing and CNV analyses besides expression studies in zebrafish and mouse and knockdown (KD) experiments using a splice blocking morpholino in zebrafish were performed to study the role of SHROOM4 during embryonic development.
Results In this study, we identified putative disease-causing SNVs and CNVs in SHROOM4 in six individuals from four families with congenital anomalies of the urinary tract and the anorectal, cardiovascular and central nervous systems (CNS). Embryonic mouse and zebrafish expression studies showed Shroom4 expression in the upper and lower urinary tract, the developing cloaca, the heart and the cerebral CNS. KD studies in zebrafish larvae revealed pronephric cysts, anomalies of the cloaca and the heart, decreased eye-to-head ratio and higher mortality compared with controls. These phenotypes could be rescued by co-injection of human wild-type SHROOM4 mRNA and morpholino.
Conclusion The identified SNVs and CNVs in affected individuals with congenital anomalies of the urinary tract, the anorectal, the cardiovascular and the central nervous systems, and subsequent embryonic mouse and zebrafish studies suggest SHROOM4 as a developmental gene for different organ systems.
- nervous system malformations
- congenital, hereditary, and neonatal diseases and abnormalities
- digestive system abnormalities
- genetic counseling
- heart defects, congenital
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. SHROOM4 variants identified in this study were submitted to ClinVar.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- nervous system malformations
- congenital, hereditary, and neonatal diseases and abnormalities
- digestive system abnormalities
- genetic counseling
- heart defects, congenital
WHAT IS ALREADY KNOWN ON THIS TOPIC
So far, missense variants or structural changes of the SHROOM4 gene have been associated with Stocco dos Santos syndrome, a neurodevelopmental disorder, but not with congenital anomalies of the urinary tract and the visceral or cardiovascular systems.
WHAT THIS STUDY ADDS
Here, exome sequencing and copy number variation analyses identified single-nucleotide variants and microdeletions in SHROOM4 in six affected individuals from four families presenting with congenital anomalies of the urinary tract and the anorectal, cardiovascular and central nervous systems. Embryonic mouse and zebrafish studies support a role of Shroom4 in the development of the respective organ systems. The observed phenotypes in zebrafish following Shroom4 knockdown resemble the human phenotype.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The present study expands the previous knowledge on the neurodevelopmental influence of SHROOM4 and suggests SHROOM4 to further play an important role in the development of the urinary tract and the anorectal and cardiovascular systems.
Introduction
SHROOM4, coding for Shroom Family Member 4, is a member of the Shroom protein family that contains a N-terminal PDZ domain, a coiled coil and a C-terminal ASD2 motif (figure 1A).1 2 Based on the domain structure, SHROOM4 protein may regulate the actin cytoskeletal architecture, which is critical for cell organisation during embryonic development.2 A wide range of cells show expression of Shroom4 during murine development, including the epithelium of the neural tube and kidney.1 2 It is further expressed in adult and fetal mouse brain structures, suggesting its implication in neural function and development.1 2

Exome sequencing and copy number variation analyses in families with congenital malformations identify variants in SHROOM4. (A) Protein domain structures are depicted (https://smart.embl.de/). The position of newly identified single-nucleotide variants of families A and B and the partial deletion of SHROOM4 in families C and D are annotated in red. Previously reported single-nucleotide variants are shown in black. (B) Pedigree with two affected individuals in family A . III-1 presented with several congenital malformations with her maternally derived X-chromosome being activated in 84% of all lymphocytes. Pedigrees of families C and D that bear microdeletions including CLCN5 and SHROOM4 are depicted. Healthy carriers of variants are highlighted with a dot. Symbols representing affected individuals are shaded with different fill for differentiating clinical conditions. Black boxes depict the congenital malformation phenotype. Striped boxes illustrate Dent’s disease. Brackets denote adoption. Triangle denotes miscarriage. (C) Amino acid sequence conservation among species of p.Glu314Lys that was altered in SHROOM4 in family A. CC, coiled coil; wk, gestational age in weeks.
So far, single-nucleotide variants (SNVs) in SHROOM4 have been associated with Stocco dos Santos syndrome (MIM: 300434). Affected individuals show developmental delay (DD), mild-to-severe intellectual disability (ID), seizures, behavioural problems, autistic features, ataxia, short stature and skeletal abnormalities (online supplemental table S1).3–7 Additionally, two individuals with moderate ID were found to have balanced X;autosome translocations with Xp11.2 breakpoints disrupting SHROOM4 (online supplemental table S1).5 Several studies using array-based comparative genomic hybridisation (CGH) in individuals with DD/ID and additional anomalies revealed microduplications, microdeletions and complex rearrangements of 0.14–8.9 Mb in size comprising chromosomal region Xp11.22 with SHROOM4 (online supplemental table S1).8–15 Two recent studies report seven SNVs in SHROOM4 found in individuals with epilepsy.16 17 Furthermore, Heide et al 18 found a nonsense variant in a fetus with corpus callosum agenesis. However, the phenotype of individuals with variations in SHROOM4 remains ill-defined.
Supplemental material
We present six individuals from four families with congenital anomalies of the urinary tract and the anorectal, cardiovascular and central nervous systems (CNS), in whom exome sequencing and copy number variation (CNV) analyses detected rare and novel SNVs in SHROOM4, and microdeletions comprising SHROOM4, respectively. Embryonic mouse and zebrafish studies suggest the additional role of Shroom4 in the development of the urinary tract and the anorectal and cardiovascular systems, in addition to the CNS.
Methods
Human subjects
DNA was extracted from blood or saliva. Saliva samples were collected using the Oragene DNA self-collection kit (following the Oragene DNA Purification Protocol for saliva samples).
Exome sequencing
Exome sequencing was conducted in family A. The family was previously described by Hilger et al.19 For enrichment of genomic DNA, the NimbleGen SeqCap EZ HumanExome Library V.2.0 enrichment kit was used. A 100 bp paired-end read protocol on an Illumina HiSeq2000 sequencer was used. Filtering of mapped target sequences and data analysis were performed using the ‘Varbank’ exome analysis pipeline (https://varbank.ccg.uni-koeln.de/varbank2/) as described previously.20 In short, variants were ranked on the basis of their probable effect on the function of the encoded protein considering evolutionary conservation among orthologues across phylogeny using ENSEMBL Genome Browser (https://www.ensembl.org/index.html) and assembled using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), as well as the in silico prediction programmes PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg/), Combined Annotation Dependent Depletion (CADD) (https://cadd.gs.washington.edu/) and MutationTaster (http://www.mutationtaster.org/). Splice site variants were evaluated using the in silico prediction tools MaxEnt (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html), NNSPLICE (https://www.fruitfly.org/seq_tools/splice.html), SSF (http://www.umd.be/searchsplicesite.html) and EX-SKIP (https://ex-skip.img.cas.cz/). Variant filtering based on population frequency was performed using population database gnomAD (https://gnomad.broadinstitute.org/) to include only rare alleles (ie, minor allele frequency <1%). Remaining variants were confirmed by Sanger sequencing. The website GeneMatcher (https://genematcher.org/) enabled the connection between researchers caring for family B.21 22
For family B, exome libraries were prepared using the Illumina TruSeq PCR-Free library preparation kit with 10 cycles of PCR, followed by enrichment with the IDT xGen Exome Research Panel V.2, with additional spike-in oligos (Integrated DNA Technologies) to capture the mitochondrial genome and dispersed genomic regions for CNV detection. Samples were sequenced to a minimum of 7 Gb of 2×125 paired-end reads for a mean of 80× average coverage or greater on the Illumina HiSeq 4000 or Illumina NovaSeq 6000. Bidirectional sequence is assembled, aligned to reference sequences based on human genome build GRCh37/UCSC hg19 and analysed using custom-developed software, RUNES and VIKING (https://www.childrensmercy.org/childrens-mercy-research-institute/research-areas/genomic-medicine-center/data-and-software-resources/). The analysis is confined to the genes of interest for the individual and with a minor allele frequency corresponding to disease incidence.
Copy number variants
Initial diagnosis in individual II-2 (family C) was performed using genome-wide oligonucleotide microarray (SurePrint G3 ISCA V.2 8×60 k, Agilent Technologies) with a mean resolution of 60 kb. For molecular karyotyping of individuals II-1 and II-2 in family C, we used the Illumina Infinium Global Screening Array-24 V.2.0 plus multidisease add-on content BeadChip. Visualisation of deletion was performed using GenomeStudio Genotyping Module V.2.0.5 (www.illumina.com/). Array analysis in family D was performed using the SurePrint G3 ISCA V.2 8×60 k (Agilent Technologies). Detected CNVs in our study were evaluated for overlapping CNVs annotated in the Database of Genomic Variants (DGV) (http://dgv.tcag.ca) and in the DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources (DECIPHER) (https://decipher.sanger.ac.uk/).
Skewed X-chromosome inactivation
Testing for skewed X-chromosome inactivation was performed in individuals II-2 and III-1. We determined the X-inactivation status at the CAG repeat of the first exon of androgen receptor locus AR (NM_000044.6). DNA was treated with methylation sensitive restriction endonuclease HpaII. Treated and untreated DNAs were amplified by PCR with FAM-fluorescence labelled HUMARA primer 1 (5′-GCTGTGAAGGTTGCTGTTCCTCAT-3′) and primer 2 (5′-TCCAGAATCTGTTCCAGAGCGTGC-3′).23 PCR products were separated on an ABI PRISM 3100 Genetic Analyzer (Thermo Fisher). The fluorescence-labelled PCR fragments were analysed with GeneMapper software (Applied Biosystems). The peak area of each fragment was quantified and compared between treated and untreated samples to determine the methylation pattern of maternally and paternally inherited X-chromosomes. Testing showed skewed X-inactivation in III-1 with her maternally derived X-chromosome being activated in 84% of all lymphocytes.
Mouse in situ hybridisation (ISH)
Mouse embryos from a wild-type (wt) SWISS background of embryonic days (E) 12.5 were dissected into PBS and fixed overnight in 4% PFA at 4°C. Embryos were processed into paraffin wax, and 5 µm sections were made using a microtome. The probe corresponds to the 3′ coding region of Shroom4 (ENSMUSG00000068270). Two primers were used to amplify a 995 bp region from mouse embryo cDNA (forward: TTGGGGCCCGAAAGAAGGTC, reverse: TTCCCTGCCATCCACATGCT), and at the reverse primer, a T7 polymerase sequence was included. The protocol of the probe generation can be found online (http://mamep.molgen.mpg.de). In vitro transcription was performed using a nucleotide mix containing digoxigenin-11-UTP (Roche). All samples were processed for ISH as described by Chotteau-Lelièvre et al 24 with minor modifications, and detection of AP activity was carried out using BM Purple (Roche). Images were obtained on a Leica M205C System with a colour camera.
Zebrafish lines and maintenance
Zebrafish were kept according to national law and to recommendations by Westerfield25 in our zebrafish facility in Bonn, Germany. Zebrafish larvae (zfl) of wt AB/TL and the transgenic Tg(wt1b:eGFP) reporter lines were obtained by natural spawning and were raised at 28°C in Danieau (30%) on a 14-hour light:10-hours dark cycle. All zebrafish experiments were performed at ≤5 days post fertilisation (dpf). To suppress pigmentation, 0.003% 1-phenyl-2-thiourea was added to the Danieau solution for respective zfl from 1 dpf onward. Staging was performed according to Kimmel et al.26
Whole-mount zebrafish ISH
The cDNA plasmid for the preparation of antisense and sense probes for shroom4 was generated by PCR from zebrafish poly-T embryonic cDNA with specific primers (forward: CATCATctgcagcgatgagatctgtgagaatgagc, reverse: CATCATggatcccagctttttacgcagactctcc). The resulting PCR products were cloned into pGEM-T Easy. Constructs were linearised by corresponding restriction enzymes, and Dig-labelled RNA was synthesised using Roche Dig labelling kit. ISH was performed following the instructions of Thisse and Thisse.27
Microinjections of morpholino oligonucleotides (MOs) and mRNA
Embryos at the one-cell to two-cell stages were pressure injected into the yolk with a splice blocking MO synthesised by GeneTools, LLC. Injections were carried out with 1.27 ng of shroom4 MO (1.8 nL/embryo) (5′-ACATTTGTGTGTTTGCTTACCTTCG-′3) and 1.27 ng of standard control (Ctrl) MO (5′-CCTCTTACCTCAGTTACAATTTATA-′3) that targets transcripts shroom4-201 (ENSDART00000111542.5) and shroom4-202 (ENSDART00000170100.3). For human mRNA rescue experiments, 75 pg of in vitro transcribed human SHROOM4 mRNA was injected into the yolk of one-cell embryos. SHROOM4 mRNA was transcribed from cDNA clone H06D041O16 (Source BioScience) containing NM_020717.4 using the mMESSAGE Machine T7 Kit (Ambion 1340M) and the Poly (A) Tailing Kit (Ambion AM1350).
Western blot analysis
Zfl were pooled into samples of 20–30 and lysed in RIPA buffer on ice with 4% protease inhibitor using a sonicator. Protein sample (50 µg) was separated by sodium dodecyl-sulfate polyacrylamide gel electrphoresis (SDS-PAGE), transferred on PVDF membranes and probed with a custom made anti-Shroom4 antibody (1:1000; polyclonal, rabbit AB2501, anti Aa 550–565, C-SERFATNLRNEIQRKK; Thermo Fisher Scientific) at 4°C overnight.
Sulforhodamine 101 (SR101) excretion assay
Excretion assay with 0.02 mM SR101 was performed on 5 dpf as previously described by our group.28
Eye-to-head ratio
Zfl were phenotyped at 4 dpf using a ZEISS Stemi508 for brightfield imaging. The time point of 4 dpf was chosen since the phenotype was most prominent. The diameters of the eyes were measured with NIS-Element Viewer software. To account for variation and growth effects, eye size was normalised to head (snout to otic vesicle).29 Zfl were anaesthetised with 0.03% tricaine and fixed in 1.25% low-melting agarose for imaging.
Phalloidin stainings
Phalloidin stainings were performed according to previous reports.30 31 shroom4 MO-injected zfl were fixed at 5 dpf in 4% PFA overnight at 4°C and subsequently washed three times for 5 min in phosphate-buffered saline (PBS) with 0.1% Tween-20 (PBST) and 5 min in PBS-Tx (2% Triton X-100). Afterwards, zfl were incubated for 2 hours in PBS-Tx to allow permeabilisation. Stainings with 2 µg/mL tetramethyl rhodamine B isothiocyanate (Sigma Aldrich, P1951) were conducted overnight at 4°C. Stained zfl were briefly washed in PBS before applying DAPI (ACDBio, RNAscope DAPI, 320858) overnight at 4°C. Zfl were washed in PBS before imaging.
Imaging
At the stages of interest embryos were analysed under a Nikon AZ100 Macro-Zoom microscope. Selected embryos were anaesthetised with 0.016% tricaine, fixed in 2% low-melting agarose and imaged by two-photon scanning fluorescence in vivo microscope (LaVision Trim-ScopeII, Imspector and ImageJ software).
Statistical analysis
Two-tailed Student’s t-test, Mantel-Cox and two-way analysis of variance test were used for analysis using GraphPad Prism V.6. Differences with a p value of<0.05 (*) were considered as being statistically significant.
Results
Exome sequencing identifies rare variants in SHROOM4
In family A, the index individual (III-1) and her maternal uncle (II-3) showed four major component features (CFs) of the VATER/VACTERL association (MIM: 192350) (figure 1B and table 1; for detailed information, see online supplemental Note). An initial CNV analysis did not detect any disease-causing CNV in III-1, II-2 and II-3 of family A.19 Skewed X-chromosome inactivation testing in the index individual (III-1) and her mother (II-2) demonstrated skewing of the X-chromosome bearing the wt allele in III-1 with her maternally derived X-chromosome being activated in 84% of all lymphocytes tested (figure 1B and table 1). Successive exome analysis prioritising X-chromosomal variants revealed a rare variant in SHROOM4 (NM_020717.4), detected in the affected individuals II-3 and III-1 and in the conducting mother (II-2) and maternal grandmother (I-2) (NM_020717.4:c.940G>A p.(Glu314Lys), ClinVar: SCV002498760), but not in the other family members I-1, II-1 and III-2 (figure 1A,B, and table 1). The p.(Glu314) glutamate is conserved among vertebrates down to Xenopus tropicalis (figure 1C). Other vertebrates exhibit the amino acid aspartate, also acidic, at this respective position. Three out of four in silico prediction programmes rate the amino acid change as potentially damaging (CADD: 20.5, MutationTaster: disease causing, SIFT: deleterious, PolyPhen-2: 0.003 (benign)). One heterozygous female carrier in 178 700 alleles (allele frequency 0.000005596) for this variant has been reported in the gnomAD online database (table 1). The variant does not reside in one of the two known functional protein domains (figure 1A). According to the standards and guidelines for the interpretation of sequence variants of the American College of Medical Genetics and Genomics (ACMG), we rated this variant as variant of uncertain significance (VUS).
Supplemental material
Genetic and phenotypical overview of affected individuals with changes in SHROOM4
Using the online tool GeneMatcher, we identified another male individual (family B, II-1) presenting with a sacral dimple, atrial septal defect, unilateral kidney dysplasia, bilateral clinodactyly of the fifth finger, left-sided single palmar crease and pes equinovarus (table 1).21 22 Additionally, he showed dysmorphic craniofacial features, gastro-oesophageal reflux, hypotonia and failure to thrive (table 1). A hemizygous novel splice site variant in SHROOM4 was identified by exome sequencing (NM_020717.4:c.3942+1G>A, ClinVar: SCV002498761), not reported in gnomAD (figure 1B and table 1). The variant resides in the essential splice-donor site, likely causing retention of intronic DNA or splicing out of exons, possibly disrupting the ASD2 motif (figure 1A).2 32 Four in silico splicing programmes (MaxEnt, NNSPLICE, SSF and EX-SKIP) predict that the nucleotide change has a 100% impact on splicing. The variant was maternally inherited (I-2) (figure 1B and table 1). The mother had a medical history of depression and seizures; furthermore, her medical history describes a cardiomegaly of unknown origin with medical reports lacking. The mother died in the first year of the child’s life. The variant was rated as VUS according to the ACMG guidelines.
Microdeletion Xp11.23p11.22 with loss of SHROOM4 and CLCN5
Individual II-1 from family C presented with congenital anomalies and neurocognitive impairment (figure 1B and table 1). He showed DD, later ID, short stature, posterior urethral valves (PUVs) and clinical characteristics of Dent’s disease. Based on the findings of subdural hygroma and retinal bleeding, shaken baby syndrome was suspected. His younger brother, II-2, presented with dysmorphic craniofacial features, DD, intraventricular haemorrhage, short stature, short limbs, failure to thrive, PUV and Dent’s disease (table 1). Array CGH analysis in individual II-2 identified a 1.07 Mb microdeletion at Xp11.23p11.22 spanning chrX:g.49,375,617–50,447,320 (hg19) (ClinVar: SCV002498762). The deletion involves eight coding genes (PAGE1, PAGE4, USP27X, CLCN5, AKAP4, CCNB3, DGKK and partially SHROOM4 (8 of 9 exons deleted, NG_011882.1)), and six microRNAs (figure 1A and online supplemental figure S1). SNP array analysis confirmed the deletion of individual II-2 and showed the same deletion in individual II-1 (figure 1B and online supplemental figure S1). Combining both array data, we were able to refine the breakpoints to chrX:g.49,369,600–50,447,320, with the 3′ breakpoint residing within chrX:g.49,174,104–49,369,600 and the 5′ breakpoint residing within chrX:g.50,447,320–50,450,462. Parental DNA for segregation analysis was not available since both brothers were adopted, with the biological parents being lost to follow-up.
Supplemental material
Individual II-4 from family D presented with congenital anomalies and neurocognitive impairment (figure 1B and table 1). He showed DD, increased muscular tone of the lower limbs, protruding eyes, retinitis pigmentosa, PUV, bilateral hydronephrosis, renal cortical microcysts, omphalocele, micropenis, cryptorchidism, inguinal hernia, unilateral single transverse palmar crease and clinical characteristics of Dent’s disease. The boy was the third born child to unrelated parents. The course of pregnancy was complicated by gestational diabetes, arterial hypertension and Grave’s disease. The oldest sister, II-1, is healthy (figure 1B). Individual II-2 was unavailable for clinical evaluation and genetic testing. The mother had a spontaneous miscarriage (II-3) at 6 weeks of gestation (figure 1B). Array CGH analysis in the affected individual, II-4, identified a 3.46 Mb microdeletion at Xp11.23p11.22 comprising chrX:g.49,375,617–52,838,206 (hg19) (ClinVar: SCV002498763) (figure 1A). This microdeletion was maternally inherited.
In accordance with the ACMG standards and guidelines for interpretation and reporting of postnatal constitutional CNVs, identified microdeletions in families C and D were rated as CNVs of uncertain clinical significance. We did not detect any relevant overlap with CNVs annotated in DGV. However, there was one individual annotated in DECIPHER with a 382 kb maternally inherited hemizygous deletion covering the first exon and a large proportion of intron 1 of SHROOM4.33 This individual (434791) was noted to have global DD. Other entries comprising very large (>50 Mb) deletions or entries lacking phenotype information were considered as not relevant.
Expression of Shroom4/shroom4 in murine embryos and zfl
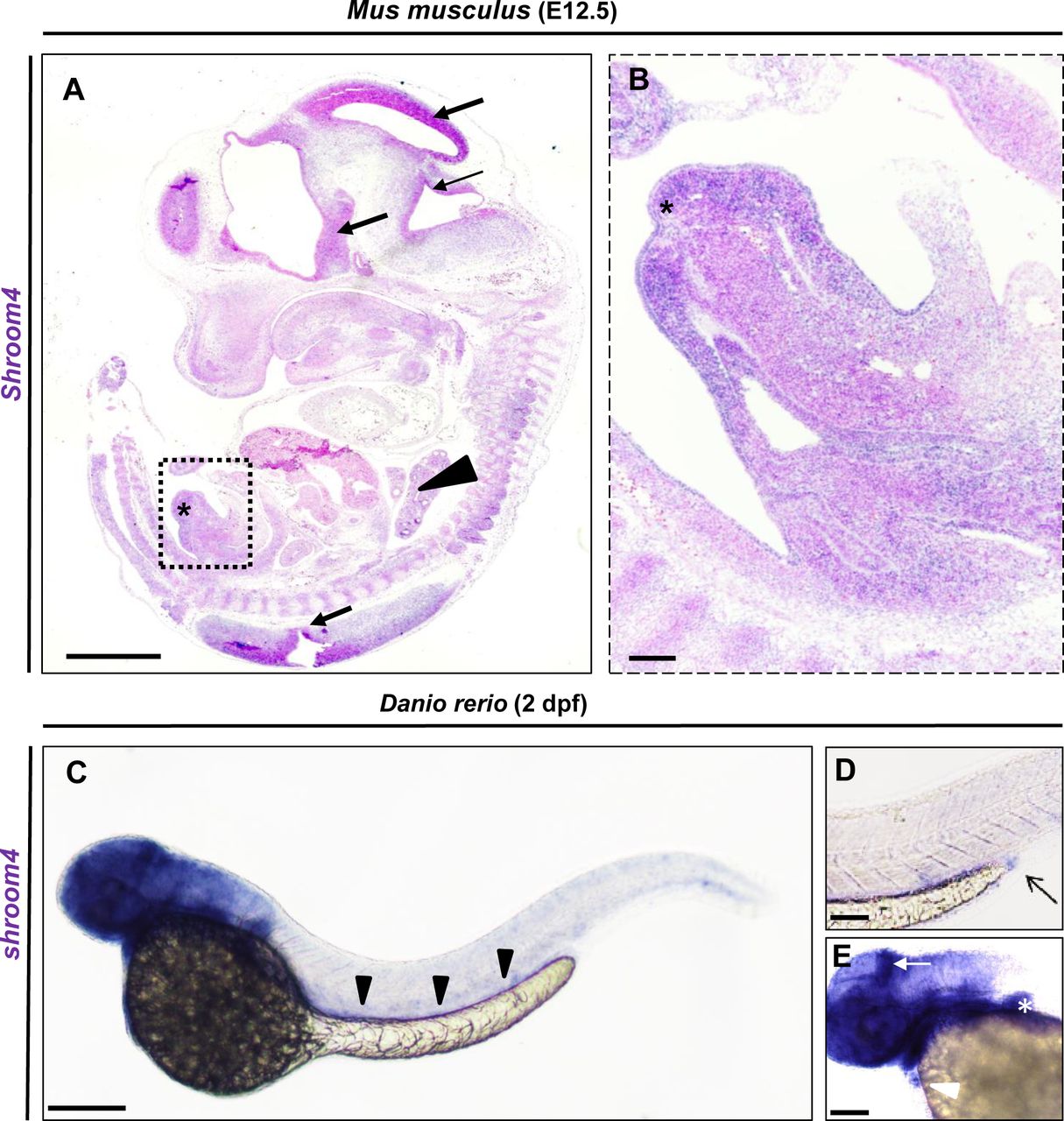
To study the expression of the canonical Shroom4 transcript (ENSMUST00000103005.10) in mouse embryos, we performed ISH studies, which showed expression in the brain and other neuronal tissues, vertebrae and genital tubercle at E12.5 (figure 2A,B). In order to further investigate the function of SHROOM4 during organ development, we used zebrafish as a model organism. BLAST analysis with human SHROOM4 identified a single zebrafish shroom4 orthologue (ENSDARG00000079900). To study the expression of shroom4 in zfl, we generated a labelled antisense RNA probe for ISH. In accordance with previous work, we detected strong expression of shroom4 in the head, brain, eyes, heart, pectoral fins, the intestine and cloacal region at 48 hours post fertilisation (hpf) (figure 2C–E) and 72 hpf (data not shown).34

Shroom4/shroom4 is expressed during early mouse and zebrafish development. (A,B) ISH with Shroom4 probe on a sagittal section of a representative E12.5 (TS21) Mus musculus. Shroom4 expression is visible (in blue) in the head and neuronal tissues (black arrows), Vertebrae (black arrowhead), and genital tubercle (asterisks). Magnification (square in A) highlights the expression in the developing genitourinary tract (B). (C) Whole-mount ISH with an anti-shroom4 probe shows the expression of shroom4 RNA (in blue) at 2 days post fertilisation in the head, brain, eyes, fins, heart, the intestine (black arrowheads) and cloacal region in Danio rerio. Sense controls did not show a staining (not shown). (D) The cloaca is highlighted by an arrow in the enlargement. (E) The expression of shroom4 is highlighted by a white arrow in the brain and by a white arrowhead in the heart in the enlargement. Positive expression in the pectoral fins is marked by the white asterisk. Scale bars represent 1 mm (A), 10 µm (B) and 100 µm (C). ISH. In situ hybridisation.
Knockdown (KD) of Shroom4 leads to reduced survival, pericardial effusion (PE), glomerular cysts, abnormal eye-to-head ratio and cloacal anomalies
The similarity of the amino acid sequences between the human SHROOM4 and zebrafish Shroom4 proteins amounts to 73%. In order to study the phenotypical effect of a Shroom4 depletion in zebrafish, we applied a KD with a splice blocking MO that targets both existing transcripts, shroom4-201 (ENSDART00000111542.5) and shroom4-202 (ENSDART00000170100.3) (online supplemental figure S2A). Efficiency of MO-induced KD was demonstrated by RT-PCR with a decrease of wt shroom4 expression and presence of an alternative band without exon 2 for transcript shroom4-201 (online supplemental figure S2B-C). Similarly, RT-PCR for transcript shroom4-202 confirmed efficiency, showing an insertion of 41 intronic bp (online supplemental figure S2B,C). No change in eef1a1 (ENSDARG00000039502) expression was detected, serving as Ctrl (online supplemental figure S2C). Western blot analysis confirmed these findings on the protein level (figure 3A). Following the KD of Shroom4, we observed reduced survival of the MO-injected zfl (26%) compared with Ctrl MO-injected zfl (76%) at 5 dpf (figure 3B). Furthermore, MO-injected zfl displayed short body length, PE, a reduced eye and head size (eye-to-head ratio), as well as glomerular cysts and pronephric dilatation (figure 3C–H). PE was found in 71% of shroom4 morphants but only in 13% of Ctrl larvae (figure 3C,D). The measured eye size was normalised to head length to account for variation in embryo size (figure 3E). This ratio was significantly lower in shroom4 MO zfl (0.37) compared with Ctrls (0.46) (figure 3F). To assess the impact of the Shroom4 KD on the kidneys and urinary tract, we used the transgenic Tg(wt1b:eGFP) fluorescent reporter line, expressing GFP in the kidney during development (figure 3G,H).35 We observed the occurrence of glomerular cysts and dilatation of the pronephric ducts at 2 dpf in 52% of with MO-injected zfl but only in 13% of Ctrls (figure 3G,H). Rescue experiments that were conducted by coinjection of human wt SHROOM4 mRNA together with shroom4 MO resulted in statistically significant increased survival (58%) (figure 3B). The observed phenotype PE in MO-injected zfl could be rescued (MO+wt RNA: 51%) (figure 3C). The reduced eye size and head length could also be rescued by coinjection with human wt SHROOM4 mRNA to a ratio of 0.42 (figure 3F). Nevertheless, the occurrence of glomerular cysts and pronephric dilatation could not be rescued (47%) (figure 3G). Overexpression of human wt SHROOM4 mRNA did not cause increased mortality or any phenotypical changes (figure 3B,C,F,G).
Supplemental material

Knockdown of Shroom4 leads to glomerular cysts, eye abnormalities and PE. (A) Western blot analysis demonstrates a protein decrease in shroom4 MO-injected zfl for Shroom4 (184 kDa, F1Q6C1), derived from transcript shroom4-201, in comparison with Ctrl MO-injected zfl. The protein product for transcript shroom4-202 is too small to be detected by Western blot analysis (11 kDa, B3DGK9). β-Actin serves as Ctrl and shows an equal amount of protein in both samples. (B) Quantification of survival (n=5); zfl injected with shroom4 MO show a significant reduction of survival rate at 5 dpf compared with Ctrl MO. Survival of shroom4 MO is significantly rescued by coinjection of human WT SHROOM4 RNA. No alterations in survival have been observed by solely injecting WT SHROOM4 RNA. (C,D) Quantification of PE (n=3) shows significant occurrence in shroom4 morphants, a phenotype, which could be rescued by co-njection of WT SHROOM4 RNA. Black arrowhead highlights PE observed in a zfl injected with shroom4 MO at 4 dpf. (E,F) Eye-to-head ratio of injected zfl at 4 dpf (n=4). Measurement of the eye (red line) and head (black line, distance between the snout to the otic vesicle) was performed as visualised (F). Injection of shroom4 MO significantly reduced eye-to-head ratio, while WT mRNA co-injection in shroom4 MO-injected zfl significantly rescues the phenotypical effect. (G,H) Zfl injected with shroom4 MO develop glomerular cysts (white arrowheads in H) and dilatation of the pronephric ducts (white asterisks in H) that could not be rescued by human WT RNA (n=4). Images from in vivo observation through fluorescence microscopy (dorsal view) in Tg(wt1b:GFP) were taken at 2 dpf. Scale bars represent 100 µm. Ctrl, control; MO, morpholino oligonucleotide; ns, nonsignificant; PE, pericardial effusion; wt, wild type; zfl, zebrafish larvae. p <0.05 (*), p <0.01 (**), p <0.001 (***)
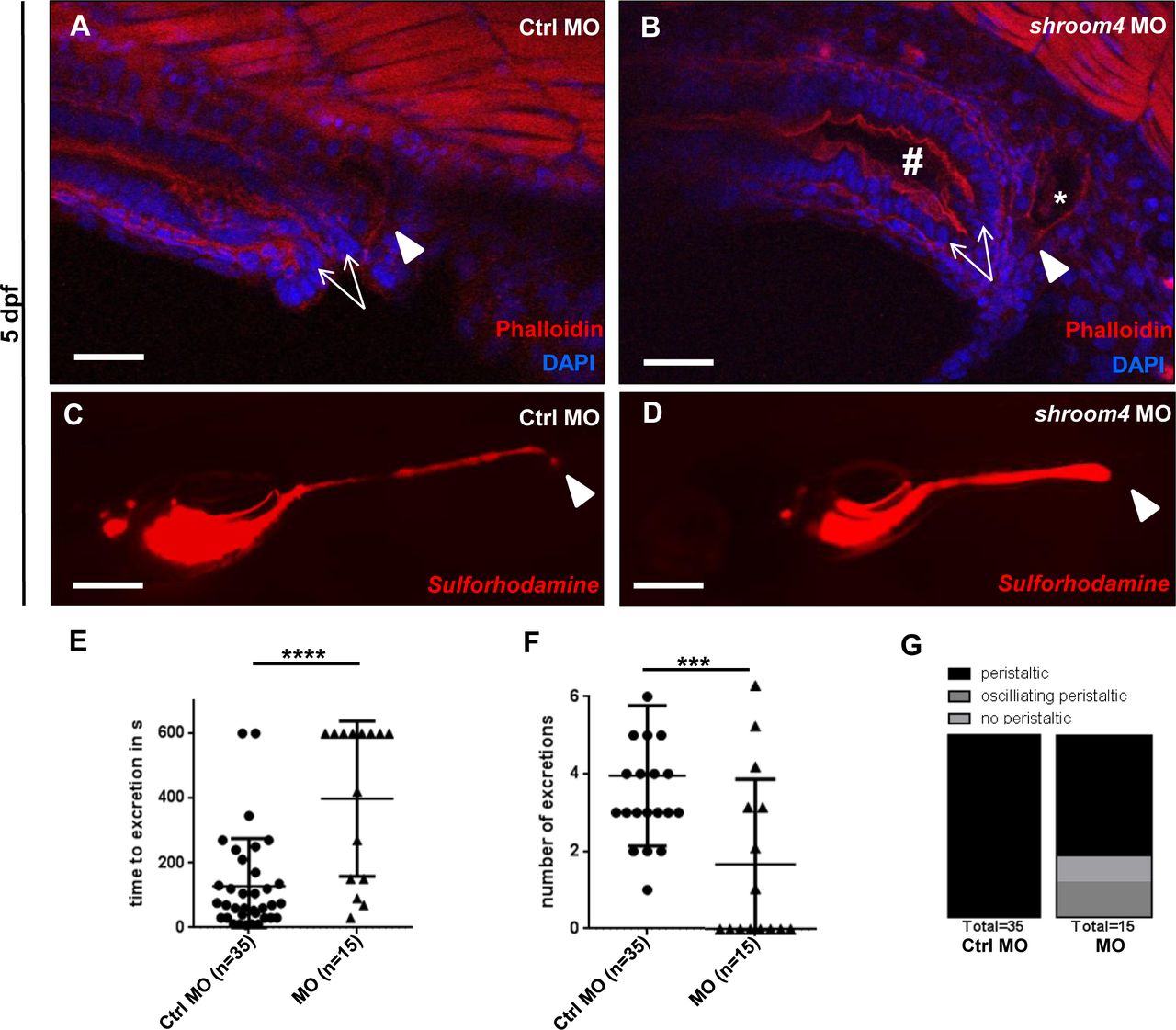
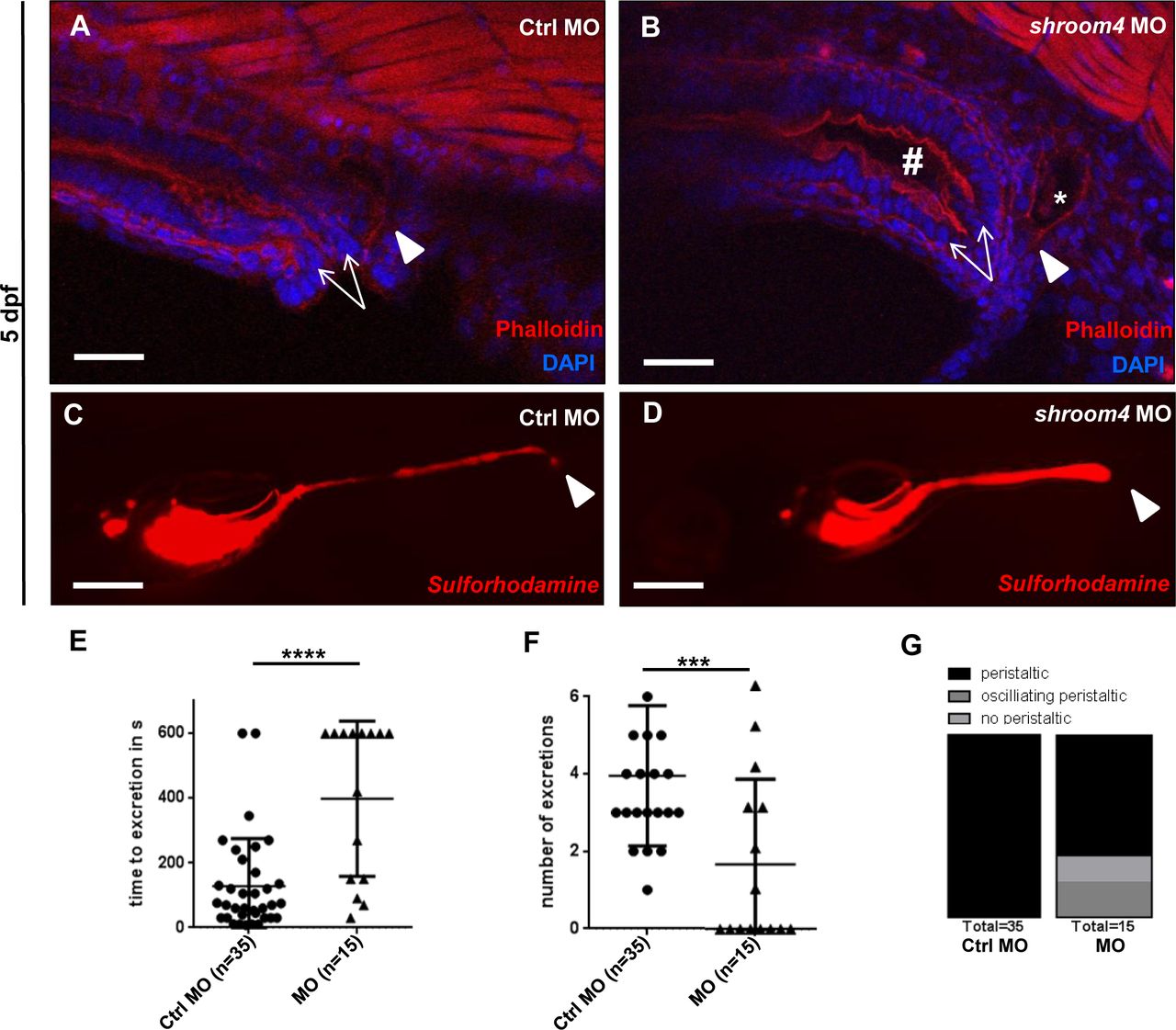
On the analogy of the observed anorectal phenotypes in family A, we were interested in studying the impact of Shroom4 depletion on cloacal development in zfl. Therefore, we applied phalloidin stainings that mark actin filaments and allow visualisation of zebrafish cloacal morphology.30 31 In comparison with Ctrls, the shroom4 MO-injected zfl exhibited a distension of the hindgut and dilated distal pronephric ducts, potentially due to distal obstruction caused by cloacal malformation (figure 4A,B). Next, we performed an SR101 excretion assay to confirm the potential hindgut anomalies in MO KD zfl. Zfl ingest SR101, a red fluorescent dye labelling the intestine, which enables examination of the opening of the cloaca and ensuing excretion of SR101 at 5 dpf. shroom4 MO-injected zfl showed a significantly prolonged time to excretion and fewer numbers of excretion (figure 4C–F). In addition, no or just oscillating peristalsis was observed in shroom4 morphants in contrast to Ctrls that all showed regular peristalsis of the intestine (figure 4G). These assays demonstrate the high abundance of cloacal opening defects in shroom4 morphants resembling the anorectal phenotype in humans.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phalloidin stainings and SR101 excretion assay show cloacal obstruction in shroom4 morphants. (A,B) Cloacal region is depicted at 5 dpf. White arrows indicate the hindgut; the white arrowheads show the distal pronephric ducts. With shroom4 MO-injected zfl display a dilated hindgut and distal pronephric morphology compared with Ctrls. (C,D) KD with shroom4 MO causes an abnormal cloacal excretion of SR101 compared with Ctrl MO-injected zfl at 5 dpf elucidated by white arrowheads (C,D). (E–G) shroom4 morphants show significantly longer time to excretion (E), significantly reduced number of excretions over time (F), and oscillating or missing peristalsis compared with Ctrls (G) (n=3). Scale bars represent 30 µm (A,B) and 100 µm (C,D). Ctrl, control; MO, morpholino oligonucleotide; SR101, sulforhodamine 101; zfl, zebrafish larvae.
Discussion
Previously, variations in SHROOM4 have been associated with Stocco dos Santos syndrome (MIM: 300434), a neurodevelopmental disorder (online supplemental table S1).3–15 36 Here, we identified genetic variations of different size affecting SHROOM4 in individuals with multiple congenital anomalies of the urinary tract, the anorectal and cardiovascular systems, and the CNS. The respective individuals presented with overlapping phenotypical features, including congenital anomalies of the urinary tract system (6/6), the CNS (4/6), the anorectum (2/6) and the cardiovascular system (2/6) (table 1). The two affected individuals of family A fulfil the clinical criteria of VATER/VACTERL association, defined by the presence of at least three of the following CFs: vertebral anomalies (V), anorectal malformations (A), cardiac defects (C), tracheo-oesophageal fistula and/or oesophageal atresia (TE), renal anomalies (R) and limb anomalies (L).19 Previously described affected individuals with ID and DD carrying variations in SHROOM4 presented with features that concern anatomical structures affected by the VATER/VACTERL spectrum (online supplemental table S1).3–6 10–14 These comprised the heart in one individual, kyphosis and scoliosis, small hands, small feet, camptodactyly and clinodactyly (online supplemental table S1). Nevertheless, kyphosis and scoliosis may be secondary to a neurogenic genesis, and the described limb anomalies are observed in other syndromes and are not specific for VATER/VACTERL association (ie, small hands and feet).
Pathogenic variants in CLCN5 are associated with Dent’s disease, a renal proximal tubulopathy, characterised by proteinuria, hypercalciuria and hyperphosphaturia, kidney stones and, in some cases, kidney failure.37 Around 8% of pathogenic variants are deletions.38 Three of the affected individuals described here carry a deletion comprising CLCN5, causing the phenotype of Dent’s disease. However, all three individuals presented also with PUV (3/3), an anatomical obstruction of the urethra not associated with Dent’s disease. Additionally, all three individuals (3/3) show neurodevelopmental disorders, also not associated with pathogenic CLCN5 variations. Our human genetic data suggest that the SHROOM4 deletions may be implicated in the formation of urinary tract anomalies and the neurodevelopmental disorders in these individuals. Interestingly, microdeletions comprising CLCN5 and SHROOM4 have been previously associated with DD, ID, growth retardation, dysmorphic craniofacial features and Dent’s disease (online supplemental table S1).9 10
The observed phenotypes in shroom4 MO KD zfl resemble somehow the phenotypical spectrum observed in the affected individuals with genetic SHROOM4 variations reported here. The KD of Shroom4 led to decreased eye diameter and head size. The observation of a reduced eye-to-head ratio within the morphant zfl suggests a growth-independent effect. This assay has been previously used as a proxy to easily observe effects on neurogenesis, migration and patterning, showing the close relation of eye size and neurodevelopmental disorders in the zebrafish model.39 Embryonic expression data in mouse embryos and zfl implicate Shroom4/shroom4 in early brain formation, implicating that the observed reduced eye-to-head ratio could represent the neurodevelopmental phenotypes in individuals with Stocco dos Santos syndrome, that is, microcephaly, ID and DD (online supplemental table S1).6 10 Further phenotypical features of shroom4 MO morphants show affection of the cardiovascular system displaying PE. However, PE might be a non-specific MO effect also observed in Ctrl zfl. Although MO-induced PE could be rescued by wt SHROOM4 mRNA coinjection, it remains uncertain if PE following Shroom4 KD resembles the cardiovascular phenotypes of families A and B.40 41 The formation of the pronephric cysts and dilated ducts in shroom4 KD zfl is consistent with the observed anomalies of the upper and lower urinary tract in the affected individuals (table 1). These also resemble the bilateral hydronephrosis caused by an anatomical obstruction due to PUV observed in individual II-4 of family D (figures 3 and 4 and table 1). The dilatation of the pronephric ducts in zfl could also be secondary to the mechanical obstruction of the observed cloacal malformation. The phalloidin staining and SR101 assay frequently visualised cloacal opening defects of the hindgut among shroom4 MO KD zfl, resembling the ARM in the affected individuals (figure 4B,D, table 1). The complete absence or only partial presence of peristalsis could be due to mechanical obstruction or possibly to a malfunction in the visceral nervous system.
The observed phenotypes PE, reduced survival and reduced eye-to-head ratio were significantly rescued by the coinjection of shroom4 MO and human wt SHROOM4 mRNA (figure 3B,C,F). These rescue experiments highlight that the observed phenotypes in shroom4 MO KD zfl, which resemble several phenotypical features of the reported families, are each specifically caused by the depletion of Shroom4 but can be reduced by coinjection of human wt SHROOM4 mRNA. While coinjection of shroom4 MO and human wt SHROOM4 mRNA resulted in a non-significant reduction of glomerular cyst formation, it still underlines the specificity of our KD for Shroom4. Together, the phenotypical changes in shroom4 MO KD zfl and the observed expression of Shroom4 in embryonic mouse tissues suggest SHROOM4 to be implicated in the development of CNS and other principal structures, that are, the urinary tract and the anorectal and cardiovascular systems. Whereas our KD experiments leading to depletion of Shroom4 resemble the genetic situation of the individuals with SHROOM4 deletions reported here, our data cannot provide evidence beyond all doubts that the missense variant of family A is indeed responsible for the respective phenotype, as we have not directly tested the variant in family A in our MO KD rescue experiments.
Hence, while additional functional studies are warranted investigating the impact of SHROOM4 SNVs, our study suggests that the developmental role of SHROOM4 might be beyond its embryonic role in CNS formation, including an additional role in the development of several other organ systems.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. SHROOM4 variants identified in this study were submitted to ClinVar.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the ethics committees of the Medical Faculty of the University of Bonn and of the institutions of respective collaborators (Lfd. Nr. 031/19). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the participating families and physicians for their contribution. Zebrafish work was supported by Bonn medical faculty zebrafish core facility. Tg(wt1b:GFP) zebrafish was provided by Christoph Englert. This study makes use of data generated by the DECIPHER community. A full list of centres which contributed to the generation of the data is available online (https://deciphergenomics.org/about/stats) and via email from contact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
HMR and GCD contributed equally.
Contributors CMK, TF, ÖY, TTL, BO and GCD performed zebrafish experiments. LS, IT, LB, TP, HT, ACH, HMR and GCD performed exome evaluation and variant analysis. IT, MS, MZ, PA, AB, FH and HMR recruited families and gathered clinical data for the study. ACH, MS, MZ, PA and PGn performed the array analysis. KH and UM conducted X-inactivation studies. PGr conducted mouse experiments. BO, HMR and GCD designed and oversaw the entire study and wrote the manuscript with CMK. GCD is responsible for the overall content as guarantor.
Funding CMK was funded by the SciMed BONFOR stipends (O-149.0120 and O-167.0021), by the German Research Foundation (DFG, KO 6579/2-1) (708037-809683, 499462148) and supported by the Biomedical Education Program (BMEP). IT and TP work was made possible by the generous gifts to Children’s Mercy Research Institute and Genomic Answers for Kids program at Children’s Mercy Kansas City. Data are available online (https://github.com/ChildrensMercyResearchInstitute/GA4K). FH was supported by the National Institutes of Health NIH (DK068306). ACH was funded by the BONFOR grant (O-149.0123) and partially funded by the Else Kröner-Fresenius-Stiftung and the Eva Luise und Horst Köhler Stiftung (project number 2019_KollegSE.04). We are grateful for a German Research Foundation (DFG) equipment grant (INST 1172/37-1 FUGG) to BO for a multiphoton microscope set-up. HR was funded by the German Research Foundation (DFG, RE 1723/4-1) and by the Else-Kröner-Fresenius-Stiftung (EKFS, 2014_A14). G.CD was funded by the BONFOR grant (O-120.0001).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.