Article Text

Abstract
Background Joubert syndrome (JS) is a recessively inherited ciliopathy characterised by congenital ocular motor apraxia (COMA), developmental delay (DD), intellectual disability, ataxia, multiorgan involvement, and a unique cerebellar and brainstem malformation. Over 40 JS-associated genes are known with a diagnostic yield of 60%–75%.
In 2018, we reported homozygous hypomorphic missense variants of the SUFU gene in two families with mild JS. Recently, heterozygous truncating SUFU variants were identified in families with dominantly inherited COMA, occasionally associated with mild DD and subtle cerebellar anomalies.
Methods We reanalysed next generation sequencing (NGS) data in two cohorts comprising 1097 probands referred for genetic testing of JS genes.
Results Heterozygous truncating and splice-site SUFU variants were detected in 22 patients from 17 families (1.5%) with strong male prevalence (86%), and in 8 asymptomatic parents. Patients presented with COMA, hypotonia, ataxia and mild DD, and only a third manifested intellectual disability of variable severity. Brain MRI showed consistent findings characterised by vermis hypoplasia, superior cerebellar dysplasia and subtle-to-mild abnormalities of the superior cerebellar peduncles. The same pattern was observed in two out of three tested asymptomatic parents.
Conclusion Heterozygous truncating or splice-site SUFU variants cause a novel neurodevelopmental syndrome encompassing COMA and mild JS, which likely represent overlapping entities. Variants can arise de novo or be inherited from a healthy parent, representing the first cause of JS with dominant inheritance and reduced penetrance. Awareness of this condition will increase the diagnostic yield of JS genetic testing, and allow appropriate counselling about prognosis, medical monitoring and recurrence risk.
- genetic variation
- central nervous system diseases
- cerebellar diseases
- congenital
- hereditary
- and neonatal diseases and abnormalities
- early diagnosis
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not Applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- genetic variation
- central nervous system diseases
- cerebellar diseases
- congenital
- hereditary
- and neonatal diseases and abnormalities
- early diagnosis
Introduction
Joubert syndrome (JS, MIM #213300) is a clinically and genetically heterogeneous ciliopathy with congenital onset, with a population-based prevalence reaching 1.7 per 100 000 in the age range 0–19 years.1 Typical neurological features include infantile hypotonia evolving into ataxia, abnormal ocular movements (mainly congenital ocular motor apraxia (COMA) and nystagmus), developmental delay, intellectual disability of variable severity, and neonatal breathing dysregulation in a subset of children. In addition, a significant proportion of patients present with involvement of other organs, such as retinal dystrophy, infantile or juvenile nephronophthisis, liver fibrosis and skeletal defects, which can manifest congenitally or later in life.2
The diagnosis of JS is confirmed neuroradiologically by the detection of a complex mid-hindbrain malformation; the ‘molar tooth sign’ (MTS) is the key element, characterised by thick, long and horizontally oriented superior cerebellar peduncles (SCPs) and often, a deep interpeduncular fossa.3 Additional features are vermis hypoplasia, upper cerebellar folial dysplasia and abnormal shape of the fourth ventricle with cranially displaced fastigium.4 Recognition of the MTS allows diagnosis at birth or even during the second trimester of pregnancy by fetal MRI.5 In JS, axonal navigation is also affected, commonly resulting in non-decussation of pyramidal tracts and efferent tracts in the SCPs.6
To date, recessively inherited variants in over 40 genes have been associated with JS, overall accounting for 60%–75% of families. All genes encode proteins implicated in the structure or function of the primary cilium, a subcellular organelle that plays key roles in embryonic development and adult tissue homoeostasis.2
SUFU encodes the major repressor of Sonic Hedgehog signalling, a key neurodevelopmental pathway mediated by the primary cilium.7–9 In 2018, we reported recessive hypomorphic missense SUFU variants as causative of JS and polydactyly in four patients from two unrelated families.10 Recently, heterozygous truncating SUFU variants were identified in 15 individuals from six families presenting with COMA, a congenital developmental disturbance of voluntary gaze characterised by the inability to initiate saccades, mainly horizontal. Of note, several individuals also had developmental delay, mild learning disability, mild ataxia and abnormal vermis and SCPs on MRI reanalysis, questioning whether a subset of COMA is actually JS.11
Here we report de novo or inherited heterozygous truncating or canonical splice site SUFU variants in 22 patients from 17 families, initially referred for genetic testing of JS-related genes due to suggestive clinical and/or neuroimaging features. As a result, we delineate a consistent neurodevelopmental syndrome encompassing the mild end of the JS spectrum, associated with presumed haploinsufficiency of the SUFU gene.
Patients and methods
To search for heterozygous truncating or canonical splice site SUFU variants, we reviewed available next generation sequencing (NGS) data in two independent large cohorts of patients referred for molecular testing of JS genes over the past 15 years. For both cohorts, ethical approval was in place to perform clinical and genetic studies, and parents had signed a written informed consent, which also included the possibility of being recontacted over the course of the project.
Patients were referred for: (1) JS diagnosis based on typical clinical features and detection of the MTS reported by the local neuroradiologist; or (2) Some clinical features of JS (hypotonia, developmental delay, COMA) without obvious evidence of a clear-cut MTS or other definitive imaging diagnosis such as tubulinopathy or Poretti-Boltshauser syndrome.
The first cohort (EU-Cohort) was recruited mainly from European countries by the Valente lab; it consists of 534 probands who underwent either NGS-based genetic testing of a custom panel of JS-causative genes (including SUFU) or clinical exome/whole exome sequencing. In this cohort, causative biallelic variants in other JS-associated genes were detected in 321 subjects (60.1%).
The second cohort (US-Cohort) was recruited mainly from the USA by the Doherty lab; it consists of 563 probands (521 with and 42 without a clear MTS). In this cohort, causative biallelic variants in other JS-associated genes had been identified in 385 subjects (68.4%).
On selection of patients carrying a heterozygous stop-gain, frameshift, or canonical splice site variant in the SUFU gene, parents were recontacted to provide detailed information about the clinical phenotype and progression over time. Available brain MRIs were carefully reviewed by at least two experts in cerebellar malformations (EuBo, FA, GEI and/or DD)
Segregation analysis was performed in all but two families. Parents found to carry the variant identified in their offspring were telephonically interviewed with regard to their developmental steps, school achievement and other potentially relevant clinical signs. Brain MRIs were performed on three reportedly asymptomatic carrier parents.
Results
We detected heterozygous truncating and canonical splice site SUFU variants in 22 patients: 14 from 10 families (1.9%) in the EU-Cohort and 8 from 7 families (1.2%) in the US-Cohort. Mean age at examination was 8 years (range 1–16), and male:female ratio was 19:3 (male 86%). A schematic of the identified variants is reported in figure 1.
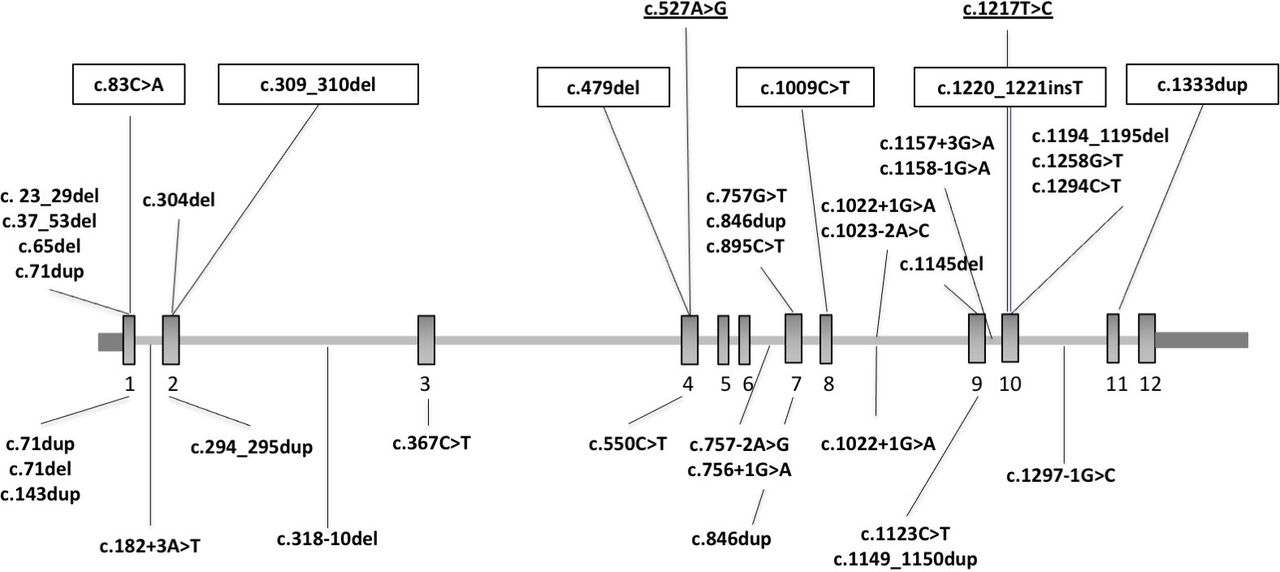
Schematic of the SUFU gene (NM_001178133.1) and variants reported so far in patients with mild JS, COMA, Gorlin syndrome or cancer. SUFU gene structure and location of reported variants. Upper panel shows heterozygous truncating and canonical splice site variants identified in the present study, as well as heterozygous LOF variants identified in patients with COMA (boxed),11 and homozygous missense variants identified in patients with JS (underlined).10 Lower panel shows heterozygous truncating and canonical splice site variants identified in patients with Gorlin syndrome or cancer. Note that three variants recurred in patients with neurodevelopmental phenotypes and in patients with cancer. COMA, congenital ocular motor apraxia; JS, Joubert syndrome.
Variants arose apparently de novo in eight probands, while they were inherited from a reportedly asymptomatic parent in 11 affected children from seven families. During the course of the study, the probands in families UW423 and UW427 were discovered to have inherited the same variant from their fathers, who were first cousins through their mothers (both obligate carriers). These two cousins reported the occurrence of isolated COMA in one sister, who was unavailable for genetic testing. In families COR572 and UW435, parents were not available for genetic testing; however, family COR572 consists of two affected biological siblings (both adopted), indicating that the variant was inherited from one of the parents; for family UW435, clinical records indicated that the proband’s mother had macrocephaly, suggesting possible maternal inheritance of the variant. Male:female ratio among the tested asymptomatic carriers was 4:4 (male 50%).
The majority of affected children presented with the same constellation of neurological features seen at the mildest end of the JS spectrum, characterised by early onset hypotonia persisting in infancy, COMA, mild motor and speech delay, and mild truncal and limb ataxia. Of these, COMA was the only invariable feature, occurring in all patients and often persisting, even if attenuated, over the years. Ataxia occurred in over 60% of the patients, while only about a third eventually manifested intellectual disability. However, when present, the degree of intellectual disability ranged from mild to severe. All children older than 5 years attended primary school, with about half of them requiring special education support. Abnormal neonatal breathing was reported only in a minority of patients. Of note, macrocephaly (either confirmed as cranial circumference above 98 percentile, or subjectively reported by parents) was present in 19 out of 22 children, while extraneurological involvement was extremely rare (table 1, online supplemental tables 1 and 2). Attention-deficit/hyperactivity disorder, autism traits, obsessive-compulsive disorder and other behavioural defects were reported in a minority of patients.
Supplemental material
Clinical and neuroimaging features of affected and asymptomatic carriers of heterozygous truncating and canonical splice site SUFU variants
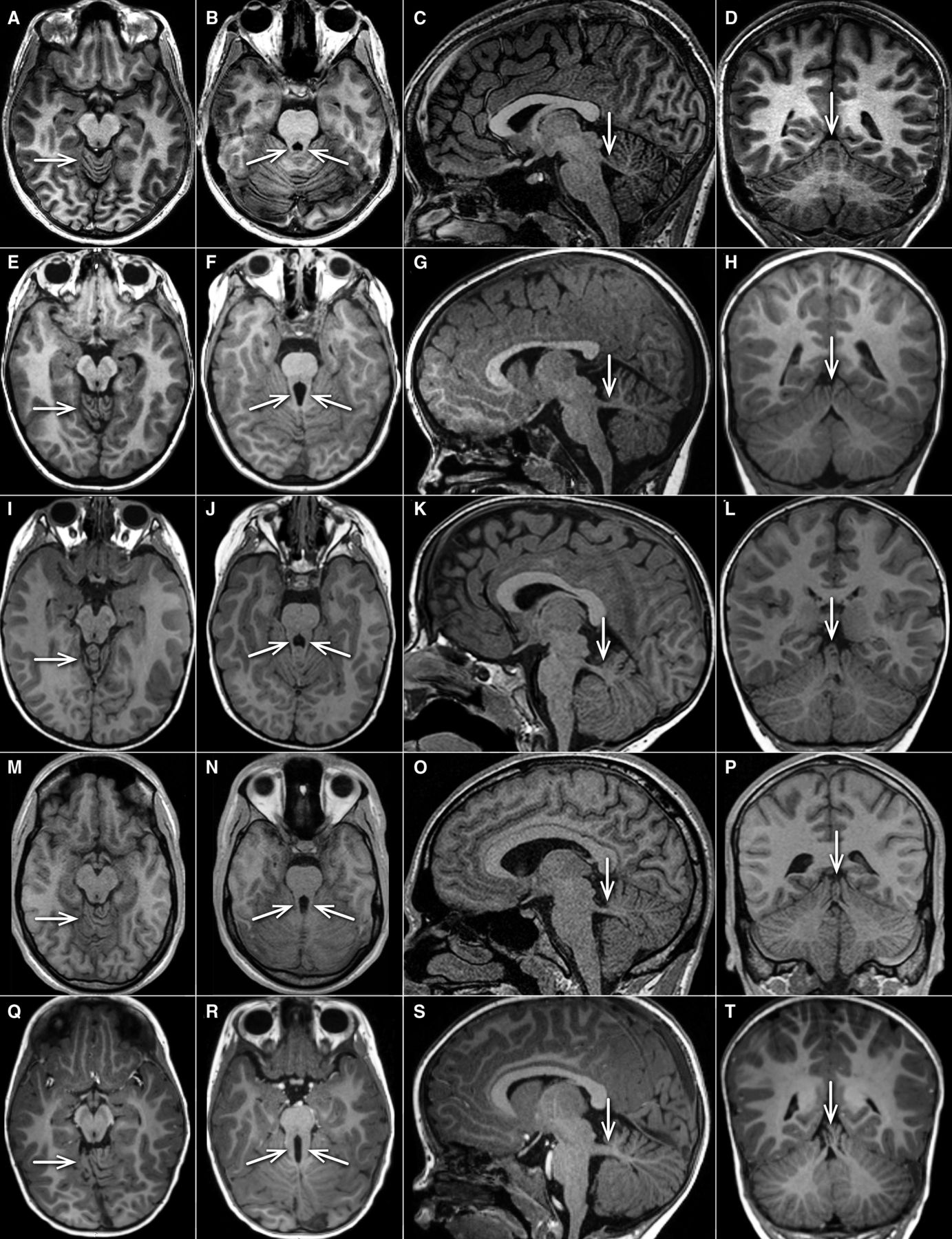
This homogenous clinical phenotype was mirrored by neuroimaging findings consistent across the whole cohort, showing a well recognisable pattern (figure 2, table 1, online supplemental tables 3 and 4). All MRIs displayed vermis hypoplasia (well seen on sagittal and coronal cuts) and superior cerebellar dysplasia (best seen on axial cuts at the level of the upper vermis), and all but one had mild but clear abnormalities of the SCPs, which were either horizontal, long and/or thick. Due to image quality (cuts too thick, not all planes available) the SCPs could not be judged in all planes in every individual. In several cases, this anomaly configured as an intermediate between normal anatomy and a typical MTS. A vermian split and a cranially displaced fastigium were common but inconsistent features, while supratentorial anomalies were not present. Of note, diffusion-tensor imaging tractography (‘fibre tracking’) failed to show evidence of disturbed axonal guidance in two affected siblings from family COR280.12

{kind=link}
{kind=link}
Representative MRIs of cerebellar vermis and SCPs in healthy control, SUFU heterozygous carriers and JS with ‘mild’ and ‘typical’ MTS. Four representative T1-weighted MRIs (arranged in horizontal rows) are shown from one asymptomatic control (10–15 years old, A–D), two individuals (0–5 years old) with SUFU LOF heterozygous variants (families COR280 and COR552) (E–H), and (I–L), one individual with JS associated due to homozygous NPHP1 deletion (15–20 years old, (M–P)), and one individual with JS associated with biallelic pathogenic AHI1 variants (0–5 years old, (Q–T)) (all unpublished). The first column (A, E, I, M, Q) shows axial views at the level of the upper cerebellum, demonstrating folial dysplasia (arrow) in all individuals except the control. The second column (B, F, J, N, R) illustrates axial views at the level of the SCPs (arrows), which are more prominent (longer, thicker)) illustrates axial views at the level of the SCPs (arrows), which are more prominent (longer, thicker) compared with normal (mild MTS in row 2–4, typical MTS in row 5). The third column (C, G, K, O, S) shows parasagittal sections demonstrating thick and horizontal SCPs in all individuals shown except the healthy control (arrow). The fourth column (D, H, L, P, T)illustrates coronal images revealing variable irregular folia and vermis splitting (arrows) in all individuals shown except the control. JB, Joubert syndrome; MTS, molar tooth sign; SCP, superior cerebellar peduncle.
Eight carrier parents were interviewed. All reported normal development, did not need extra help in school and denied any neurological issues, with the exception of one mother who reported mild clumsiness and ataxia during school age. We were able to perform brain MRI in three healthy carriers. Notably, images from two carriers were indistinguishable from those of the patients, showing the same characteristic constellation of features (figure 2, table 1), while in a third carrier brain imaging was normal.
Discussion
To date, biallelic hypomorphic SUFU variants have been associated with mild JS,10 and heterozygous truncating variants with COMA.11 Here we describe heterozygous truncating and canonical splice site SUFU variants in 22 patients either diagnosed with JS (defined by the presence of the MTS on brain imaging) or showing some clinical and/or imaging features suggestive of JS, and further delineate the clinical and neuroradiological spectrum associated with SUFU haploinsufficiency.
In infancy, the typical symptoms of COMA, hypotonia, developmental and speech delay are often indistinguishable from those seen in patients with genetically confirmed JS, as well as in other forms of congenital ataxia, such as Poretti-Boltshauser syndrome due to LAMA1 pathogenic variants.13 Yet, the long-term prognosis of the SUFU-related condition is overall favourable compared with classic JS: about 60% of our patients became mildly ataxic, and fewer than half manifested intellectual disability or required school support. Another important observation is the lack of retinal, kidney, liver or skeletal involvement typical of JS; in our cohort, which includes several patients already in their second decade, the phenotype is purely neurological, with additional features rarely observed in single patients. A possible clinical clue for SUFU-related conditions is represented by macrocephaly of variable degree; however, it must be noted that a large head circumference has been reported as a common finding in young children with JS, often resolving with age.14 Interestingly, of the formerly reported 15 patients diagnosed with COMA and carrying heterozygous truncating SUFU variants, nearly all presented overlapping neurological features, including developmental and/or speech delay, early onset truncal and gait ataxia and, to a lesser extent, learning disability, indicating that these patients and the patients reported here are affected by the same neurodevelopmental condition related to SUFU haploinsufficiency.11
Further supporting this observation, the imaging pattern is also highly consistent: indeed, all patients in this study as well as those reported by Schröder et al 11 showed a combination of vermis hypoplasia, superior cerebellar folial dysplasia and abnormalities of the SCPs, which variably appear long, thick and horizontal. This pattern often results in a ‘mild MTS’ appearance, similar to what has already been reported in patients with JS carrying pathogenic variants in other genes, including NPHP1, CPLANE1, CBY1 and FAM149B1 (figure 2M–P).15–18 Yet, in our experience, this ‘mild MTS’ can often remain unrecognised by clinical neuroradiologists, resulting in misdiagnosis. It is worth mentioning that adequate imaging technique is required to allow correct identification of this malformation, including images in all three planes, and a slice thickness not exceeding 2–3 mm.19 Experience with fibre tracking in patients with SUFU variants is very limited, and axon guidance defects, which are well documented in JS, need to be evaluated in future studies.12
We were able to interview eight reportedly asymptomatic carrier parents. They all denied COMA, developmental delay and any neurological issues, with the exception of one who complained of mild ataxia/clumsiness in infancy. The proportion of asymptomatic carriers in our cohort seems higher than that reported by Schröder et al, yet it must be noted that these subjects were not clinically examined and therefore subtle signs of ataxia or abnormal ocular movement cannot be ruled out with certainty. However, somewhat surprisingly, four of the five clinically asymptomatic carriers who underwent brain MRI (three from the present study and two from the former COMA study) presented the same imaging pattern as observed in affected individuals, suggesting that the penetrance of the brain malformation is higher than that of the clinical phenotype. Further studies on clinically asymptomatic carriers will be required to confirm this observation.
While over 40 genes have been associated with JS, variants in five ‘common genes’ (AHI1, CC2D2A, CEP290, CPLANE1, TMEM67) cause almost half of JS cases, each gene accounting for 5%–10% families.20 Heterozygous truncating and canonical splice site SUFU variants seem to account for 1%–2% of patients who had been referred for JS genetic testing, either because they received a definite diagnosis of JS or because they presented clinical and/or imaging features that are part of the JS phenotype. Thus, we recommend that SUFU should be included in any diagnostic sequencing panel for JS, as well as for isolated COMA. Moreover, it is important to stress that SUFU haploinsufficiency represents the first ‘non-recessively inherited’ cause of JS. In a diagnostic setting, this implies that the variant filtering pipeline should be optimised to include heterozygous SUFU variants.
The genetic diagnosis of SUFU haploinsufficiency bears major consequences for genetic counselling. Our data show that less than half of SUFU variants arose apparently de novo, while the rest were inherited from a clinically asymptomatic carrier parent. Thus, while the recurrence risk for JS is usually 25%, here the recurrence risk in future pregnancies is going to be up to 50% when a parent carries the variant while, for apparently de novo variants, the empirical recurrence risk can be set at about 1%, to take into account the possibility of germinal mosaicism in one parent.
The occurrence of germline heterozygous truncating SUFU variants has been found to predispose to a variety of tumours, such as basal cell carcinoma, meningioma and cerebellar medulloblastoma,21–25 and in some cases to Gorlin (nevoid basal cell carcinoma) syndrome, characterised by the occurrence of several basal cell carcinomas and other cancers at a young age (<20 years), variably associated with developmental and skeletal abnormalities (figure 1).26 27 So far, no patients with the SUFU-related neurodevelopmental phenotype have presented with any obvious signs of Gorlin syndrome, nor have they developed cancer. Similarly, COMA, cerebellar dysplasia and other neurological signs typical of JS have never been reported in patients with Gorlin syndrome, although motor delays have been occasionally described in some patients. While it seems that mild JS and Gorlin syndrome represent SUFU-related allelic conditions, nevertheless genetic counselling should take into account the possibility of an increased risk for cancer and discuss the opportunity of appropriate surveillance, as tumours may also occur in adulthood.28
The mechanisms underlying such phenotypical diversity associated with SUFU loss of function variants, as well as their reduced penetrance, remain to be explained. We can speculate that additional molecular mechanisms, such as a mutational burden involving heterozygous hypomorphic variants in other JS or cancer genes, or second hits within specific cell types may be implicated in the development of either phenotype.
The human SUFU gene is extremely intolerant to truncating variants and generally intolerant to variation of all types. The gnomAD database reports only three subjects carrying heterozygous truncating variants out of ~1 40 000 individuals tested (pLI Score=1). This extremely low frequency of ‘unaffected carriers’ in the general population is not unexpected, given that JS is a very rare disease and SUFU loss of function variants only account for about 1%–2% cases. Moreover, missense variants are present in gnomAD at significantly lower frequency than expected (observed/expected ratio 0.68) with very few homozygotes. Of note, the four previously published patients with JS carrying homozygous missense SUFU variants presented with a mild clinical and neuroradiological phenotype which closely resembles that associated with SUFU haploinsufficiency, in association with polydactyly.10 This suggests that the mode of inheritance of SUFU-related disorders strongly depends on the pathogenic impact of variants, insofar truncating and canonical splice site variants are associated with dominant inheritance, while hypomorphic missense variants require both mutated alleles to manifest, thus acting in a recessive manner. Further studies are needed to establish whether certain missense variants may cause similar protein disfunction as truncating variants, thus causing a neurodevelopmental phenotype in the heterozygous state.
Gene-phenotype correlations have been clearly established for some of the the most common genetic forms of JS, which are of great help to establish appropriate surveillance as well as management and therapeutic strategies. For instance, CEP290-related JS is consistently associated with retinal and renal involvement, while TMEM67-related JS is highly correlated with liver fibrosis with or without coloboma.20 29 30 Here, we define another strong gene-phenotype correlation between heterozygous SUFU truncating variants with a mild, purely neurological JS phenotype.
In our cohort, 19 out of 22 (86%) SUFU manifesting carriers were male. This skewed male prevalence in affected individuals is not observed in global JS cohorts that have a roughly equal sex distribution (55% male in both the European and US cohorts). Interestingly, the proportion of SUFU heterozygous men was 4 out of 8 (50%) among tested asymptomatic carriers and 4 out of 10 (40%), if we also consider obligate gene carriers. By including the two additional potential carriers, who are both female (the sister of UW423-1 and UW427-1 reported with COMA, and the mother of UW435-3 reported with macrocephaly), the male proportion among carriers could even drop to 25%. In the study by Schröder et al, 10 out of 13 (77%) manifesting carriers were male, while the only two clinically unaffected carriers were both female, in line with our observation.11 Such preponderance of male sex in manifesting versus non-manifesting carriers is intriguing, suggesting that sex-related factors may affect the penetrance of SUFU variants.
We conclude that heterozygous SUFU variants must be recognised as causative of a novel, dominantly inherited neurodevelopmental syndrome encompassing COMA and mild JS, with a well recognisable imaging counterpart frequently not recognised as the MTS by clinical radiologists. The expanded phenotypical spectrum described here will aid in diagnosis and drive appropriate genetic testing, medical management, and counselling about prognosis and recurrence risk.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not Applicable.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Collaborators University of Washington Center for Mendelian Genomics (UW-CMG) group: M J Bamshad, S M Leal, D A Nickerson, P Anderson, T J Bacus, E E Blue, K Brower, K J Buckingham, J X Chong, D Cornejo Sánchez, C P Davis, C J Davis, C D Frazar, K Gomeztagle-Burgess, W W Gordon, M Horike-Pyne, J R Hurless, G P Jarvik, E Johanson, J T Kolar, C T Marvin, S McGee, D J McGoldrick, B Mekonnen, P M Nielsen, K Patterson, A Radhakrishnan, M A Richardson, G T Roote, E L Ryke, I Schrauwen, K M Shively, J D Smith, M Tackett, G Wang, J M Weiss, M M Wheeler, Q Yi and X Zhang. UW-CMG performed part of exome sequencing and initial data analysis.
Contributors VS, EuBo and EMV planned the study; VS, FD, DD, EuBo and EMV collected the data; VS, FD, YHC, AKC, HL, AM and UW-CMG performed exome sequencing, molecular or bioinformatic analyses; DD, FA, GEI and EuBo reviewed MRI exams of the recruited patients; VS, FD and SN performed statistical analyses; VS, EuBo, FA, DD and EMV wrote the first draft of the manuscript, tables and figures; JCD, JB, RoBa, EnBe, ReBo, CC, SD, JF, MF, SG, VL, FM, NM, LM, CVM, RR, WMS, SabSig, SabSil, KS, GV, MW and GZ clinically diagnosed and followed up the patients. EMV is responsible for the overall content of the manuscript acting as guarantor. All the authors revised the manuscript for important intellectual content and approved the final version.
Funding JS research in the Valente Lab is supported by grants from the Italian Ministry of Health (Ricerca Corrente 2021, Ricerca Finalizzata RF-2019-12369368), the Italian Ministry of University and Research (grant Dipartimenti di Eccellenza to the Department of Molecular Medicine, University of Pavia), Telethon Foundation - Italy (grant GGP20070), Fondazione Pierfranco and Luisa Mariani (PADAPORT project). JS research in the Doherty Lab is supported by grants from NIH (R01HD100730, R01NS064077), as well as support from the NIH-funded UW Intellectual and Developmental Disabilities Research Center (U54HD083091 PI Michael Guralnick, Genetics Core and sub-project 6849 to DD, and P50HD103524 PI Michael Guralnick, Genetics Core) and UW Pediatric Department funds. GZ and EnBe are members of the European Reference Network for Rare Neurological Disorders - ERN-RND -Project ID No 739510.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.