Article Text

Abstract
Background A large number of new causative and risk genes for amyotrophic lateral sclerosis (ALS) have been identified mostly in patients of European ancestry. In contrast, we know relatively little regarding the genetics of ALS in other ethnic populations. This study aims to provide a comprehensive analysis of the genetics of ALS in an unprecedented large cohort of Chinese mainland population and correlate with the clinical features of rare variants carriers.
Methods A total of 1587 patients, including 64 familial ALS (FALS) and 1523 sporadic ALS (SALS), and 1866 in-house controls were analysed by whole-exome sequencing and/or testing for G4C2 repeats in C9orf72. Forty-one ALS-associated genes were analysed.
Findings 155 patients, including 26 (40.6%) FALS and 129 (8.5%) SALS, carrying rare pathogenic/likely pathogenic (P/LP) variants of ALS causative genes were identified. SOD1 was the most common mutated gene, followed by C9orf72, FUS, NEK1, TARDBP and TBK1. By burden analysis, rare variants in SOD1, FUS and TARDBP contributed to the collective risk for ALS (p<2.5e-6) at the gene level, but at the allelic level TARDBP p.Gly294Val and FUS p.Arg521Cys and p.Arg521His were the most important single variants causing ALS. Clinically, P/LP variants in TARDBP and C9orf72 were associated with poor prognosis, in FUS linked with younger age of onset, and C9orf72 repeats tended to affect cognition.
Conclusions Our data provide essential information for understanding the genetic and clinical features of ALS in China and for optimal design of genetic testing and evaluation of disease prognosis.
- genetics
- genetic variation
- neurodegenerative diseases
- medical
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Amyotrophic lateral sclerosis (ALS) is one of the most common incurable neurodegenerative diseases that primarily affect the motor neurons, eventually resulting in progressive paralysis and death 3–5 years after disease onset.1 Approximately 5%–10% of patients with ALS have a family history (FALS), while the remaining cases are sporadic (SALS).2
To date, variants in more than 120 genes have been reported to be implicated in ALS (https://alsod.ac.uk). Variants in ~40 genes have been shown to cause or significantly increase the risk of ALS, although the pathogenicity of variants in some of the genes remains to be validated. Among them, C9ORF72, SOD1, FUS and TARDBP are the most common causative genes, accounting for 54.5% of FALS and 7.5% of SALS in Caucasians.3 Genetic studies of ALS in other populations have been reported, but largely in a small number of ALS genes and in small-sized cohorts. Comprehensive genetic studies of large ALS cohorts with unbiased approaches, such as whole-exome sequencing (WES), are limited.
In the current study, using repeat-primed PCR (RP-PCR) and WES, we studied a total of 1587 patients with ALS and 1866 inhouse controls from a cohort of Chinese population, which represents one-fifth of the world’s population, to characterise the genetic and clinical spectrum of ALS in Chinese population and provide essential information for ALS research and clinical practice. The study has the following aims: (1) systematically identify pathogenic and likely pathogenic (P/LP) (for causative genes) or deleterious (for new, needing to be confirmed or risk genes) variants of 41 known ALS-associated genes in an unprecedented large cohort of Chinese patients with ALS, summarise and compare the mutation frequencies of FALS and SALS, and perform gene/allele-based burden analyses to reveal whether rare variants of these genes contribute collectively to ALS; and (2) investigate the genotype–phenotype correlation in Chinese patients with ALS, which will benefit genetic counselling, early diagnosis intervention and prognostic assessment.
Methods
Subjects
Based on the El Escorial criteria for ALS,4 a total of 1587 patients with clinically definite and probable ALS admitted to the Department of Neurology of West China Hospital between December 2010 and June 2019 were recruited for this study. According to a previous study,5 patients with an identifiable family history of ALS among first-degree, second-degree or third-degree relatives were classified as ‘familial’ (FALS); otherwise patients were classified as ‘sporadic’ (SALS). For FALS cases, only the proband was analysed in the current study. Demographic and clinical data were collected. ALS that occurred in patients less than 45 years of age was considered as early-onset ALS. The Revised ALS Functional Rating Scale (ALSFRS-R) was applied to assess disease severity. According to a previous study,6 the progression rate was defined as: (48−‘baseline’ ALSFRS-R)/time from onset to ‘baseline’ (months). If the ALSFRS-R decline was more than 0.5 per month, it was classified as fast progression. As described in our previous studies,7 8 cognition and frontal lobe functions were assessed using the Addenbrooke’s Cognitive Examination-Revised and the Frontal Assessment Battery tests, respectively. A total of 1866 inhouse Chinese controls whose mean age was 57.9±9.0 years and had neither a medical nor family history of neurodegenerative disorders were included to assess the collective risk of rare variants for each candidate gene. All subjects who participated in the study provided informed consent prior to their participation. Genomic DNA was collected from peripheral blood leucocytes using standard phenol-chloroform procedures.
Genetic analysis
Genetics analysis, including WES and RP-PCR for testing G4C2 repeats of C9orf72, data analysis, variant annotation, and haplotype analysis of special P/LP variants identified in more than one patient have been described in the online supplemental methods or in our previous studies.9 10 According to the Amyotrophic Lateral Sclerosis Online Database (https://alsod.ac.uk/), 41 ALS causative/associated genes with varying degrees of genetic evidence involved in the study were classified into two categories, including 16 definitive ALS genes (group I, GI, or causative genes) and 25 new, needing to be validated or risk genes (group II, GII) (online supplemental table S1).
Supplemental material
Variant identification and burden analysis
Considering the low incidence of ALS, we defined variants with minor allele frequency in the Genome Aggregation Database-East Asian (GnomAD-EAS) less than 0.0001 as rare variants. For the GI genes, we classified variants as P/LP, variants of uncertain significance (VUS), and likely benign or benign according to the American College of Medical Genetics (ACMG) recommendations.11 For the GII genes, we classified variants as deleterious or non-deleterious variants according to five functional prediction tools. The methods used for variant identification are described in detail in the online supplemental methods. The sequencing kernel association test (SKAT) and Fisher’s exact test were performed on the gene/allelic basis for genetic investigations into the collective risk of rare variants in 40 genes for ALS (except C9orf72). Rare variants obtained from ALS by WES were assigned to the case group, and rare variants from 1866 Chinese controls from WES were used as the control group (figure 1).

Flow chart of the study. ACMG, American College of Medical Genetics; ALS, amyotrophic lateral sclerosis; BWA: Burrows-Wheeler Aligner; GATK: The Genome Analysis Toolkit; GI, group I, causative genes; GII, group II, new, needing to be confirmed or risk genes; GnomAD-EAS, Genome Aggregation Database-East Asian; IGV: Integrative Genomics Viewer; P/LP, pathogenic or likely pathogenic; RP-PCR, repeat-primed PCR; SKAT, sequencing kernel association test; VUS, variants of uncertain significance.
Statistical analysis
Comparison of continuous data was made using Student’s t-test. χ2 tests were used to compare categorical variables between the two groups. Kaplan-Meier curves were performed to graphically compare survival between subgroups. A two-tailed p<0.05 was considered statistically significant. Statistical analysis was performed using SPSS V.25.0. A Bonferroni correction for multiple comparisons was performed if necessary.
Results
Patient demographics and data set overview
Overall, 1587 patients with ALS, including 64 (4.0%) patients with FALS, were included in the study. Most patients (81.5%) came from Sichuan Province (in South West China; figure 2A and online supplemental table S2) and 96.4% of them were Han Chinese (online supplemental table S3). In our cohort, the mean age of onset was 53.9±11.7 years and the peak age of onset was 40–70 years, which accounts for up to 80% of all patients. At the time of analysis, 982 patients (61.9%) died; the median survival time from symptom onset to death was 40.9 years (95% CI 38.7 to 43.1) (table 1).

Regional distribution and genetic characteristics of patients with ALS in the study. (A) Distribution of patients with ALS in the study. Most of them came from Southwest China. (B) Type and proportion of rare variants identified in the study. (C) Mutation frequencies of each ALS causative gene in FALS and proportion of rare variants of each ALS causative gene in all the identified rare variants in FALS. (D) Mutation frequencies of each ALS causative gene in SALS and proportion of rare variants of each ALS causative gene in all the identified rare variants in SALS. ALS, amyotrophic lateral sclerosis; FALS, familial ALS; SALS, sporadic ALS; VUS, variants of uncertain significance.
Demographic features of patients with amyotrophic lateral sclerosis in the study
Genetic characteristics
Rare variants of causative genes in ALS
A total of 95 P/LP variants in ALS causative genes were identified in 155 patients, including 26 (40.6%) with FALS and 129 (8.6%) with SALS; the most frequent was missense (figure 2B–D, online supplemental table S4). Interestingly, none of one in the 155 patients carried two or more P/LP variants in the GI genes. The most frequent causative gene in FALS was SOD1 (21.9%), followed by FUS (6.3%), TARDBP (3.1%) and VCP (3.1%). In SALS, mutations in SOD1 were the most frequent (1.9%), followed by C9orf72 (1.31%), NEK1 (1.12%), FUS (0.98%), TARDBP (0.85%) and TBK1 (0.66%). In addition, variants defined as VUS were found in 1.6% of FALS and 3.5% of SALS, respectively (figure 2C,D, online supplemental table S14).
Supplemental material
Burden analysis
Although a large number of genetic studies for ALS have been performed in patients of European ancestry, we know relatively little regarding the genetics of ALS in Chinese, especially for the GII genes. To determine whether rare variants in a single gene were enriched in ALS cases (ALS-associated) or deleted in ALS cases (ALS-protective), we evaluated the distribution of rare variants, damaging (P/LP and deleterious) and non-damaging (VUS and non-deleterious) variants in the ALS cohort and controls from inhouse controls by SKAT (online supplemental table S6). In the causative genes, we found rare P/LP variants in SOD1, FUS, TARDBP, OPTN and UBQLN2 passed the exome-wide significance (p<2.5E-6) (table 2). Interestingly, rare P/LP variants in two genes, TBK1 and NEK1, identified as ALS causative genes by whole-exome analyses and gene burden analyses,12 13 were also enriched in ALS from our cohort, and the p value approached significance (p=0.008 and p=0.002, respectively). In the GII genes, rare damaging variants in SPG11 were significantly fewer in ALS than in controls (p<1.0E-7), which might be explained in part by the observation that rare damaging variants in SPG11 contribute to the development of the recessive form of ALS.14 However, rare variants in other GII genes were not enriched in ALS cases by gene-level burden analyses. Detailed information of rare variants identified in patients and inhouse controls is shown in online supplemental tables S14,S15.
Supplemental material
Burden analysis of rare variants (MAF <0.0001) in ALS-associated genes among 1587 patients with ALS and 1866 inhouse controls
In order to explore which rare P/LP variants in the GI genes might contribute to the risk of ALS, the distribution of minor allele frequencies of rare P/LP variants in the GI genes between patients and controls was compared by standard Fisher’s exact test by two-stage analysis. In the initial stage, rare P/LP variants in each gene from 1587 cases and 1866 inhouse controls were analysed; only G4C2 repeats in C9orf72 and TARDBP p.Gly294Val passed significant difference (online supplemental table S5). In the second stage, 1587 cases and controls from GnomAD-EAS (n=9977) were included; two additional pathogenicity variants, FUS p.Arg521Cys and FUS p.Arg521His, also exceeded significance (online supplemental table S5).
Genotype–phenotype analysis
General features of patients with rare P/LP variants in ALS causative genes
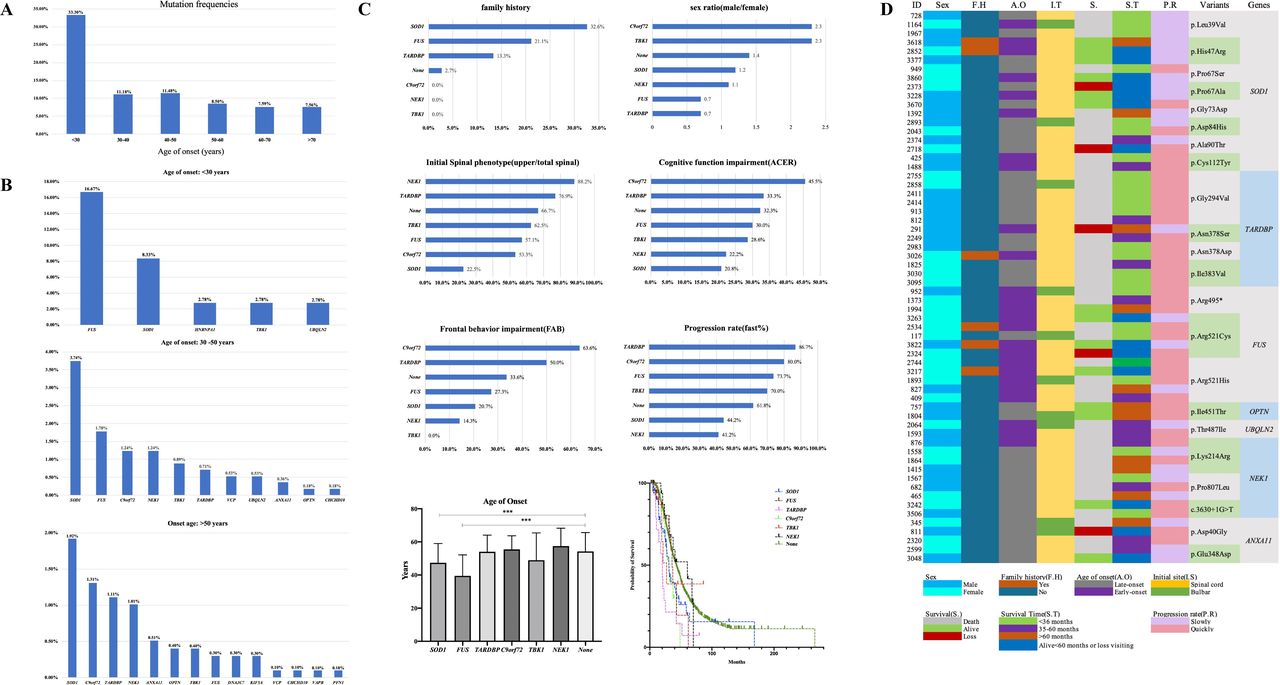
Among the 155 patients carrying rare P/LP variants, 33.3% (12 of 36) of patients aged <30 years carried P/LP variant, which was much higher than that of patients aged >30 years (figure 3A). According to mutation frequencies of patients from different groups of age of onset, the mutation spectrum of three groups of age of onset, namely <30 years, 30–50 years and >50 years, was analysed. Variants in FUS were the most frequent in the group of patients aged <30 years (16.7%), while it was SOD1 in the groups of patients aged 30–50 years and >50 years (3.7% and 1.9%, respectively) (figure 3B).

Mutation spectrum according to age of onset in patients with ALS and the genotype and phenotype correlation in ALS causative genes. (A) Mutation frequencies of patients in different groups of age of onset. (B) Ranking of frequencies of rare P/LP variants in ALS causative genes in patients whose age of onset was less than 30 years (n=36, total patients; n=12, patients with P/LP variants), more than 30 years but less than 50 years (n=562, total patients; n=64, patients with P/LP variants), and more than 50 years (n=989, total patients; n=79, patients with P/LP variants). (C) Genotype and phenotype correlation of six most frequent mutated genes, including SOD1 (n=43), FUS (n=19), TARDBP (n=15), C9orf72 (n=20), TBK1 (n=10) and NEK1 (n=17). None means patients without rare variants of the GI genes (n=1376). The phenotype, including family history, sex ratio, initial spinal symptoms, cognitive functional impairment, frontal behaviour impairment, progression rate, age of onset and median survival time were analysed. For age of onset, it was 47.4±11.6 years in patients with SOD1 variants, 39.5±12.7 years in patients with FUS variants, 54.0±10.0 years in patients with TARDBP variants, 55.5±8.2 years in patients with C9orf72 G4C2 repeats, 49.0±16.5 years in patients with TBK1 variants, 57.5±10.8 years in patients with NEK1 variants and 54.2±11.4 years in patients without rare variants of the GI genes. Significant differences were found between patients with FUS variants and without rare variants of the GI genes (p=3.0E-8) and between patients with SOD1 variants and without rare variants of the GI genes (p=0.0001). For median survival time, it was 30.5 (95% CI 25.1 to 35.9) months in patients with SOD1 variants, 35.8 (95% CI 31.5 to 40.0) months in patients with FUS variants, 20.0 (95% CI 15.2 to 24.9) months in patients with TARDBP variants, 30.9 (95% CI 25.4 to 36.5) months in patients with C9orf72 G4C2 repeats, 26.9 (95% CI 16.8 to 37.0) months in patients with TBK1 variants, 60.5 (95% CI 25.0 to 95.9) months in patients with NEK1 variants and 42.6 (95% CI 40.0 to 45.3) months in patients without rare variants of the GI genes. Significant differences were found between patients with TARDBP variants and without rare variants of the GI genes (p=6.0E-5) and between patients with C9orf72 G4C2 repeats and without rare variants of the GI genes (p=0.012) (***p<0.001). (D) Clinical characteristics of rare special P/LP variants in ALS causative genes identified in more than one patient. ACER, Addenbrooke’s Cognitive Examination-Revised; ALS, amyotrophic lateral sclerosis; FAB, Frontal Assessment Battery; GI, group I; P/LP, pathogenic/likely pathogenic.
Genotype–phenotype correlation of patients with P/LP variants in ALS causative genes
A total of 155 patients carried rare P/LP variants of 16 causative genes; the genotype–phenotype correlation is presented in detail in online supplemental table S6. Among them, more than 10 patients with rare P/LP variants were identified in SOD1, C9orf72, FUS, NEK1, TARDBP and TBK1, respectively, all of which were responsible for 7.8% (124 of 1587) of all patients and 80% (124 of 155) of patients carrying P/LP variants of ALS causative genes. Thus, the phenotype of these six genes was analysed further (figure 3C).
Most patients with SOD1 P/LP variants had a positive family history, indicating strong penetrance of mutations in SOD1. There were more male in patients with C9orf72 and TBK1 variants, but more female in patients with FUS and TARDBP variants. FUS variants presented with an early age of onset (compared with patients without rare variants of the GI genes, p=3.0E-8; figure 3C). Except for SOD1, weakness or muscular atrophy of the upper limb was usually first involved in patients carrying the other five causative gene variants. Cognitive and frontal behaviour impairments were usually seen in patients carrying the C9orf72 and/or TARDBP variants, but relatively less in SOD1, NEK1 or TBK1. Patients carrying TARDBP, C9orf72 and FUS variants presented a rapid progression, but those carrying NEK1 variants showed a slow progression. The much shorter median survival time in patients carrying TARDBP and C9orf72 variants was usually less than 36 months, but that of patients carrying NEK1 variants was more than 60 months.
Genotype–phenotype correlation among patients with variants from varying degrees of genetic evidence
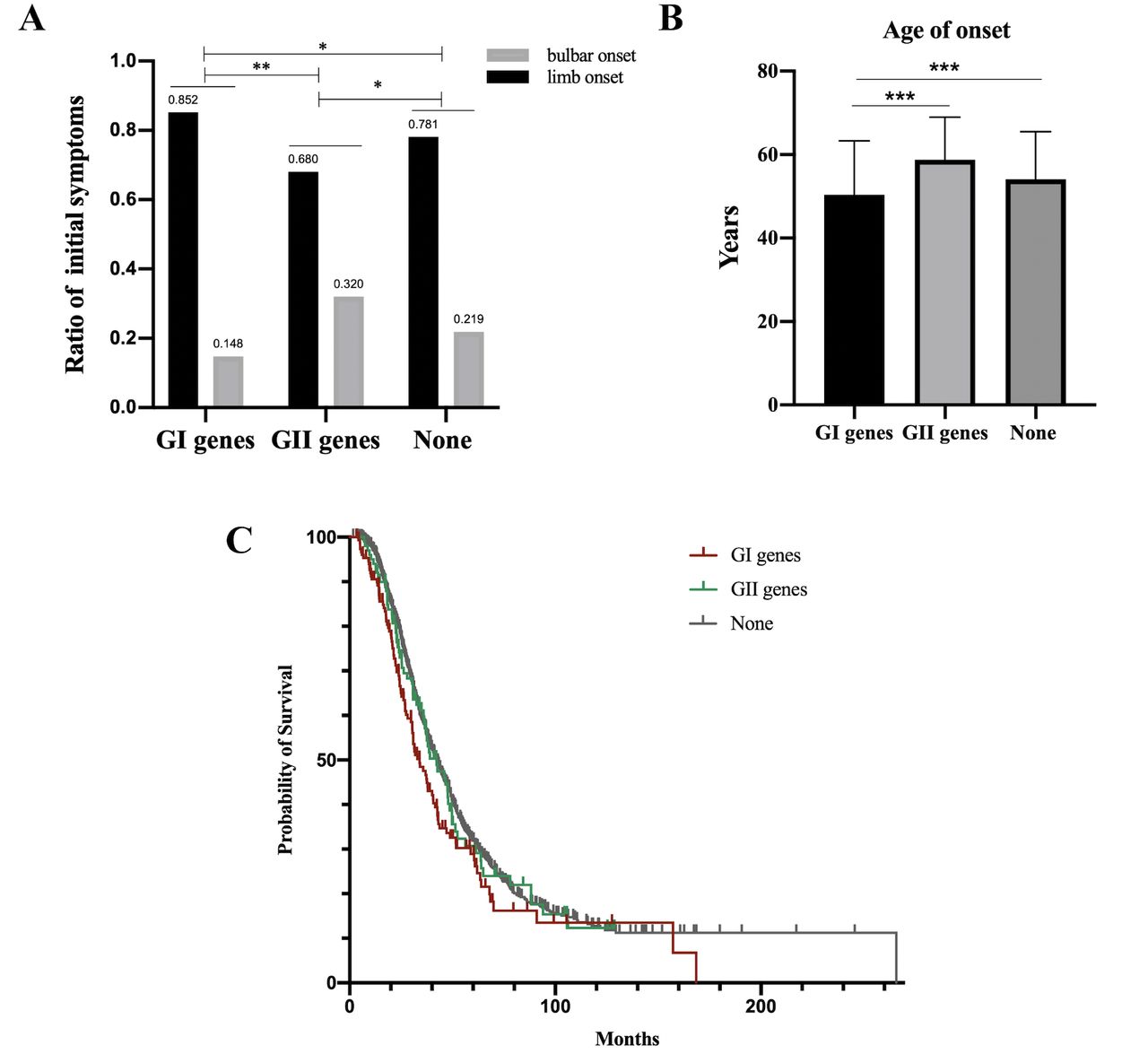
In the current study, 155 patients had rare P/LP variant of GI, 100 patients with rare deleterious variant of GII and 1276 patients without rare variants of GI and GII (56 patients with variants of VUS in GI were not analysed). Compared with patients without P/LP variants of GI and deleterious variants of GII, much higher proportion of limb onset as the initial symptom (85% vs 78%, p=0.042), much younger age of onset (50.4 years vs 54.1 years, p<0.001) and much shorter median survival time (34.0 months vs 42.6 months, p=0.011) in patients with P/LP variants of GI (figure 4A–C), and much higher proportion of bulbar onset as the initial symptom (32% vs 22%, p=0.020), were observed in patients with damaging variants of GII; however, there were no differences in the duration of diagnostic delay, ALSFRS-R at baseline and disease progression rate among these three groups (online supplemental figure S1A–C). In addition, no differences were found in cognitive function impairment and frontal behaviour impairment between patients with rare P/LP variants of GI and patients without P/LP variants of GI (online supplemental table S6). The genotype–phenotype correlation of patients with rare damaging variant of GII is described in detail in online supplemental table S7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of initial symptoms, age of onset and median survival time among patients with rare P/LP variants in the GI genes (n=155), rare deleterious variants in the GII genes (n=100) and without rare damaging variants in the GI and GII genes (n=1276). (A) Ratio of patients presenting limb onset or bulbar onset (*p<0.05, **p<0.01). (B) The mean age of onset was 50.4±12.9 years in patients with rare P/LP variants in the GI genes, 55.8±12.0 years in patients with rare deleterious variants in the GII genes and 54.1±11.4 years in patients without rare damaging variants in the GI and GII genes. The mean age of onset in patients with rare P/LP variants in the GI genes was younger than that in patients with rare deleterious variants in the GII genes (***p<0.001) and in patients without rare damaging variants in the GI and GII genes (***p<0.001). (C) The median survival time was 34.0 (95% CI 28.4 to 39.6) months in patients with rare P/LP variants in the GI genes, 42.3 (95% CI 33.5 to 51.1) months in patients with rare deleterious variants in the GII genes and 42.6 (95% CI 39.9 to 45.3) months in patients without rare damaging variants in the GI and GII genes. Significant difference was found between patients with rare P/LP variants in the GI genes and without rare damaging variants in the GI and GII genes (p=0.011). GI, group I; GII, group II; P/LP, pathogenic/likely pathogenic.
Genotype–phenotype correlation and haplotype analysis for special pathogenic variants of ALS causative genes identified in more than one patient
Twenty-two rare P/LP variants in seven GI genes were identified in more than one patient. The detailed features of the patients are shown in figure 3D. Patients carrying SOD1 p.Cys112Tyr, TARDBP p.Gly294Val, TARDBP p.Asn378Asp, TARDBP p.Ile383Val, FUS p.Arg495* and FUS p.Arg521His presented a rapid progression and/or short survival duration, but patients carrying SOD1 p.His47Arg, SOD1p.Pro67Ala and ANAX11 p.Glu348Asp presented a slow progression and longer survival duration. Haplotype analysis for special pathogenic variants identified in more than two patients revealed that the patients with the same variants shared the same haplotypes (online supplemental tables S8–S12), suggesting that these variants might be derived from founder events. However, most of the probands denied family history of ALS or mimic ALS, suggesting reduced penetrance of these mutants.
Significantly, p. Gly294Val of TARDBP, as the most common mutation in our cohort, was found in six unrelated patients who presented late onset, faster progression and shorter survival duration (<36 months from onset) (figure 3D). Interestingly, until now, this variant has only been reported in eight Caucasian patients with ALS, especially of Italian origin,15 but not found in Chinese.16 Haplotype analysis for five of them with this variant suggested they shared a haplotype interval of 397.5 kb, suggesting a common ancestral origin. Further functional experiments from immunocytochemistry (online supplemental figure S2A) and flow cytometry analysis (online supplemental figure S2B) supported that p. Gly294Val of TARDBP was pathogenic.
More than one rare variant of GI and GII genes: evidence of oligogenic inheritance
Cumulated studies proposed the possibility that multiple interacting genetic variants might enhance the risk of developing the disease or modify the disease phenotypes in neurodegenerative diseases.17 Interestingly, a total of 50 patients with more than one rare variant of GI and GII genes were identified (online supplemental table S13). Among them, 5 patients (7.8% in FALS) reported a family history of ALS, but 45 patients (3.0% in SALS) were sporadic. Although the age of onset in patients with more than one variant in the GI genes tends to be much younger than in patients with only one rare P/LP variants in the GI genes (48.9±14.3 years vs 50.8±12.6 years), no significance in age of onset, progression rate and median survival time was found between patients with more than one variant in the GI genes and only one rare P/LP variants in the GI genes (online supplemental figure S2).
Discussion
In the current study, we determined the genetic spectrum of 41 ALS-related genes and the genotype–phenotype correlation based on a large Chinese ALS cohort. Furthermore, for the first time, we systematically analysed the collective risk of rare P/LP or deleterious variants in these genes, which led to a better understanding of their pathogenicity in ALS.
To date, several genetic studies have comprehensively analysed the mutation spectrum of ALS using next generation sequencing (NGS) methods in European patients,5 18–21 providing important information for developing guidelines for genetic testing for ALS in clinical practice. In Asia, especially in China, which contributes a fifth of the world’s population, only two NGS studies on a limited sample size of patients with ALS16 22 have been performed to analyse the mutation spectrum of ALS-associated genes (45 and 268 patients with ALS involved, respectively). The small-sized studies prevented the clarification of genetic spectrum and the genotype–phenotype correlation. In addition, the clinical features of the consecutive patients involved in this study, including the proportion of FALS, sex ratio, initial symptoms, mean age of onset and median survival time, were consistent with other studies from Chinese population,22 23 although some of which were slightly different from patients from Caucasian populations (eg, the proportion of patients with FALS was 1.2%–2.7% in Chinese23 (4.0% in our cohort), but 5%–10% in Caucasians2; and the mean age of onset was 54.4 years old in a larger Chinese ALS cohort24 (53.9 years old in our cohort), but 62.1–66.3 years old in Caucasians25). Therefore, it was reasonable that the ALS cohort involved in this study could be a typical representative of the Chinese ALS population, and these differences of clinical features between Chinese and Caucasian patients indicated it is necessary to unearth the genetic features from other ethnicities.
In the present study, we comprehensively analysed the mutation spectrum of known causative and risk or needing to be confirmed ALS genes in Chinese patients with ALS, which were classified into two categories (GI and GII). We found the mutation frequencies of the 16 GI genes were as high as 40.6% and 8.6% in FALS and SALS, respectively, which were consistent with those reviewed in previous studies.23 26 According to our comprehensive analysis, we suggest a genetic screening strategy in Chinese patients with ALS: in FALS, the following genes should at least be analysed, SOD1 (21.9%), FUS (6.3%) TARDBP (3.1%) and VCP (3.1%), which account for 34.4% of patients, and in SALS, SOD1 (1.90%), C9orf72 (1.31%), NEK1 (1.12%), FUS (0.98%), TARDBP (0.85%) and TBK1 (0.66%), which account for about 6.8% of patients. Compared with mutation frequencies in patients with ALS of European ancestry,27 SOD1 is the most common causative gene in Chinese patients with FALS and SALS.26 Surprisingly, G4C2 repeat expansion in C9orf72 is the second common causative gene for SALS in our cohort, which was substantially higher than that in other Chinese cohorts (1.31% vs 0%–0.3%).28 29 However, no patients with C9orf72 G4C2 repeats had reported family history of ALS in this study, but up to 40% of C9orf72 positive FALS from Europe and the USA were identified,30 which might be partly accounted by deficient awareness of ALS in Chinese populations in the last few decades and the age-dependent penetrance of C9orf72 G4C2 repeats.30 The mutation carriers of C9orf72 G4C2 repeats from our cohort were suggested to share a common founder of European ancestry,31 indicating that it is an ideal candidate cohort for prospective studies investigating the role of C9orf72 in the Chinese population due to its age-dependent penetrance,30 and the upcoming targeted therapies for C9orf72 repeat will also benefit a subset of Chinese patients with ALS. In addition, more than 33% of patients whose age of onset was <30 years carried P/LP variants, whereas the mutation frequencies (about 11% or 8%) presented plummeting in patients whose age of onset was >30 years, which supported the hypothesis that the number of multistep process of ALS (usually six-step process)25 will be reduced in patients with ALS with genetic mutations compared with those without mutations.32 According to our findings, varied mutation frequency and spectrum among different groups of age of onset suggest that patients who are younger than 30 years should regularly undergo genetic testing to clarify the aetiology since high mutation frequencies especially for FUS were seen. Moreover, our finding also suggested that ageing and environmental factors play a more important role than genetic factors in elderly patients with ALS. Therefore, these findings might contribute to drafting a genetic scanning strategy for Chinese patients with ALS.
Our study further clarifies the correlation between genotypes and phenotypes of ALS in the Chinese population. Generally, significantly earlier age of onset, faster progression and shorter median survival time were found in patients carrying P/LP variants than in patients without P/LP variants, which was consistent with other studies and suggests that causative genetic variants are a poor predictor of outcome of ALS.1 31 However, we did not find much more proportion of patients beginning with bulbar onset in patients with the GI gene variants compared with patients without GI/GII variants because the former were generally considered a more ‘severe’ phenotype. Nearly half of patients with variants of the three genes (75 patients out of 155 patients with the GI gene variants), namely SOD1, TARDBP and NEK1, variants which will always be linked with spinal onset (as shown in our study; online supplemental table S6), might contribute to the current finding. Although phenotypic pleiotropy of ALS genes was suggested, genotype–phenotype correlation analyses further determined the main clinical manifestations of each gene. As such, patients with SOD1 variants tend to first involve lower limb. Mutations in FUS and TARDBP were associated with an earlier age of onset, faster progression and shorter survival, but a reverse relationship was found in patients with variants of NEK1 and ANXA11. Most patients with P/LP variants of C9orf72 likely presented with cognitive and frontal behaviour impairments, but relatively less in SOD1. Importantly, more clear and sharing phenotypes of each P/LP variant were characterised in our study, such as p.Leu39Val, p.His47Arg and p.Pro67Ala in SOD1, which were associated with slower progression, and p.Cys112Tyr in SOD1, p.Gly294Val in TARDBP and p.Arg521His in FUS, which were associated with faster progression. Because all of the patients came from different families and most shared variants were suggested to come from a founder effect based on our haplotype analysis, our results suggest that these causative genetic variants strongly modify disease phenotypes.
Our study further supports the findings that the most common variants in Caucasians, p.Ala4Val of SOD1 and p.Arg521Cys of FUS, are associated with faster progression,33 34 although the former was found in only one late-onset patient whose survival duration was about 14 months. However, the most common variants of TARDBP, p.Ala382Thr, reported in Caucasian patients with ALS,35 and p.Met337Val, reported in Chinese from Eastern China,16 were not found in our cohort. Another variant, p.Gly294Val, of TARDBP was first reported in Chinese and was the most common variant in our cohort. Its pathogenicity was previously suggested by experimental evidence from patient-derived induced pluripotent stem cells36 and is supported by our functional experiments (online supplemental figure S3). Our haplotype analysis was also consistent with a common ancestral origin reported in five Italian and Moroccan patients with ALS sharing a haplotype interval of 1.4 Mb.15 However, whether patients with this variant identified in our cohorts and Caucasians came from a founder effect was unknown due to differences in polymorphisms used for haplotype analysis. In any case, our results suggested it is necessary to explore the genetics of patients with ALS from different ethnicities, even from different regions in the same ethnicity, although some patients might share a few P/LP variants.
Since 2014, 12 new ALS causative/risk genes have been discovered (shown in online supplemental table S1), namely CHCHD10, TBK1, NEK1, ANXA11, KIF5A, DNAJC7, MATR3, TUBA4A, CCNF, TIA1, GLT8D1 and CYLD; only the former six genes were classified as ALS causative genes due to strong evidence (https://alsod.ac.uk/). The remaining 6 genes and other 19 ALS risk genes require replication or resolution of conflicting evidence or have not been well studied in Chinese population. However, we did not find significant enrichment of rare variants by burden analysis at the gene level, which did not support its pathogenicity in ALS. However, the results should be interpreted with caution. In our analysis, consensus definition for P/LP or damaging variants in patients with ALS and inhouse controls was performed; however, the pathogenicity predicted to be deleterious variants needs to be confirmed in future studies. In addition, aetiological heterogeneity, incomplete penetrance, late-onset diseases or limited sample size make it difficult to detect the associations between rare variants and the disease in case–control studies. As shown in the results, rare P/LP variants did not significantly enrich in each causative gene, except for SOD1, FUS, TARDBP, OPTN and UBQLN2.
The current study represents a comprehensive and systematic screening of ALS-associated genes in an unprecedented large cohort of patients with ALS from West China; however, it has some limitations. First, we focused on the mutation spectrum and conducted genotype–phenotype correlation analyses in the GI genes, but the pathogenicity of some GII genes in ALS still needs to be confirmed in future studies. Second, we only analysed the clinical and genetic architectures of patients with putative rare P/LP variants in the coding region of ALS-causative genes, but not of those with VUS. Although the pathogenicity of variants was determined according to the ACMG standard, their pathogenicity needs to be confirmed in more functional studies. Variants in non-coding regions, such as regulatory elements, which may play important roles in ALS, were not analysed in the current study.
Conclusion
We conducted the most systematic investigation of the clinical and genetic architectures in consecutive patients of a large ALS population from West China. The identified mutation spectrum of ALS is beneficial in drafting genetic scanning strategies, especially for patients with FALS and those with an age of onset younger than 30 years. The genotype–phenotype correlation analysis provided an approach for precision diagnosis ,intervention and prognosis assessment.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Approval was obtained from the Ethics Committee of West China Hospital of Sichuan University (approval number 2015-236). Informed consent was obtained from all subjects.
Acknowledgments
The authors appreciate all cohort individuals and their families for their participation in this study. The authors thank Professor Fen-Biao Gao, University of Massachusetts Medical School, for helpful discussions and comments.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Y-PC, S-HY, Q-QW and BC contributed equally.
Contributors Y-PC: design, execution, DNA extraction, patients’ enrolment, data analysis, writing. SY: execution, DNA extraction, controls’ enrolment, data analysis. Q-QW: execution, patients’ enrolment, clinical data collection. BC: design, execution, patients’ enrolment, clinical data collection, data analysis. XG: blood sample collection, DNA extraction, data analysis. WS, BZ, X-PC, YW, JY, MS: execution, patients’ enrolment. MS, F-FL: execution, data analysis. YH, RO, LZ, KL, J-YL, X-RX, ZJ, JL, YX, KC, FF, Y-YC, S-RL, TH, X-QY, X-YG, HL, QH, NS: clinical data, blood sample collection. Y-FC, C-YL, J-PL, P-LP: data analysis. Q-QZ: DNA extraction. HS: conception, design, organisation, review and critique.
Funding This study was supported by the National Key Research and Development Program of China (grant no. 2016YFC0901504), the National Natural Science Fund of China (grant no. 81871000, 81701249 and 81971188), the Science and Technology Bureau Fund of Sichuan Province (no. 2019YFS0216), Program for Entrepreneurial and Innovative Leading Talents of Guangzhou, China (grant no. CXLJTD-201603), Project of Academician Workstation at KingMed Diagnostics, Guangzhou, China (grant no. 2017B090904030), and the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (grant no. 2019HXFH046 and ZYJC18038).
Map disclaimer The inclusion of any map (including the depiction of any boundaries therein), or of any geographic or locational reference, does not imply the expression of any opinion whatsoever on the part of BMJ concerning the legal status of any country, territory, jurisdiction or area or of its authorities. Any such expression remains solely that of the relevant source and is not endorsed by BMJ. Maps are provided without any warranty of any kind, either express or implied.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.