Article Text

Abstract
Background Progressive cavitating leukoencephalopathy (PCL) is thought to result from mutations in nuclear genes affecting mitochondrial function and energy metabolism. To date, mutations in two subunits of complex I, NDUFS1 and NDUFV1, have been reported to be related to PCL.
Methods Patients underwent clinical examinations, brain MRI, skin biopsy and muscle biopsy. Whole-genome or whole-exome sequencing was performed on the index patients from two unrelated families with PCL. The effects of the mutations were examined through complementation of the NDUFV2 mutation by cDNA expression.
Results The common clinical features of the patients in this study were recurring episodes of acute or subacute developmental regression that appeared in the first years of life, followed by gradual remissions and prolonged periods of stability. MRI showed leukoencephalopathy with multiple cavities. Three novel NDUFV2 missense mutations were identified in these families. Complex I deficiency was confirmed in affected individuals’ fibroblasts and a muscle biopsy. Functional and structural analyses revealed that these mutations affect the structural stability and function of the NDUFV2 protein, indicating that defective NDUFV2 function is responsible for the phenotypes in these individuals.
Conclusions Here, we report the clinical presentations, neuroimaging and molecular and functional analyses of novel mutations in NDUFV2 in two sibling pairs of two Chinese families presenting with PCL. We hereby expand the knowledge on the clinical phenotypes associated with mutations in NDUFV2 and the genotypes causative for PCL.
- central nervous system diseases
- diagnosis
- genotype
- neurodegenerative diseases
- phenotype
Data availability statement
Data are available on reasonable request. The data that support the findings of this study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Leukoencephalopathy is seen in disorders of toxic, acquired vascular or infectious origin as well as in inherited disorders. Inherited leukoencephalopathies are a large group of heterogeneous disorders that result in white matter abnormalities.1 2 Cavitating leukoencephalopathy associated with mitochondrial complex I deficiency was first described in 1999,3 while progressive cavitating leukoencephalopathy (PCL) was first proposed in 2005 and is thought to result from mutations in nuclear genes affecting mitochondrial function and energy metabolism.4 In 2011, Ferreira and colleagues first reported that the NDUFS1 gene was related to PCL and associated with respiratory chain complex I (Co I) deficiency. The study concluded that the clinical and neuroradiological entity of PCL is a syndrome strongly related to mitochondrial respiratory chain enzyme deficiency.5 Subsequently, NDUFV1 and other genes involved in mitochondrial function and energy metabolism were also reported to be related to PCL.6–10 Both NDUFS1 and NDUFV1 encode mitochondrial Co I subunits.
Co I (NADH: quinone oxidoreductase) is by far the largest and most complicated enzyme of the respiratory chain and is composed of more than 40 subunits, 14 central subunits and up to 32 accessory subunits, 7 of which are encoded by mitochondrial DNA and the remaining by nuclear DNA (nDNA).5 6 11 Co I can be subdivided into three functional modules: the electron input (N) module that oxidises NADH, the P module that pumps protons across the membrane and the electron output (Q) module that reduces quinone. The N module consists of the subunits encoded by the nDNA genes NDUFV1, NDUFV2 and NDUFS1.6 11
NDUFV2 is a highly conserved core subunit of Co I and functions in the transfer of electrons from NADH to the respiratory chain. To date, only six likely pathogenic mutations in NDUFV2 have been reported, of which two mutations (c.427C>T, c.580G>A) result in Leigh syndrome,12 two mutations (c.120+5_120+8delGTAA, c.669_670insG) lead to hypertrophic cardiomyopathy and encephalopathy13–17 and Leigh syndrome,17 while the other two mutations (c.86C>T, c.626A>G) are likely related to Parkinson’s disease.18 19 To date, no leukoencephalopathy phenotype has been reported.
Here, we report the clinical findings, neuroimaging and molecular and functional analyses of novel mutations in NDUFV2 in four Chinese patients (two sets of siblings) presenting with PCL. Thus, we expand the knowledge on the clinical phenotypes of NDUFV2 and the genotypes of PCL.
Methods
Participants
Sibling pairs from two families (F-1 II-1 and II-2; F-2 II-1 and II-2) participated in this study, and their pedigrees are shown in figure 1. The families were recruited by the Department of Neurology, Beijing Children’s Hospital, Capital Medical University and by the Department of Pediatrics, Peking University First Hospital (China).
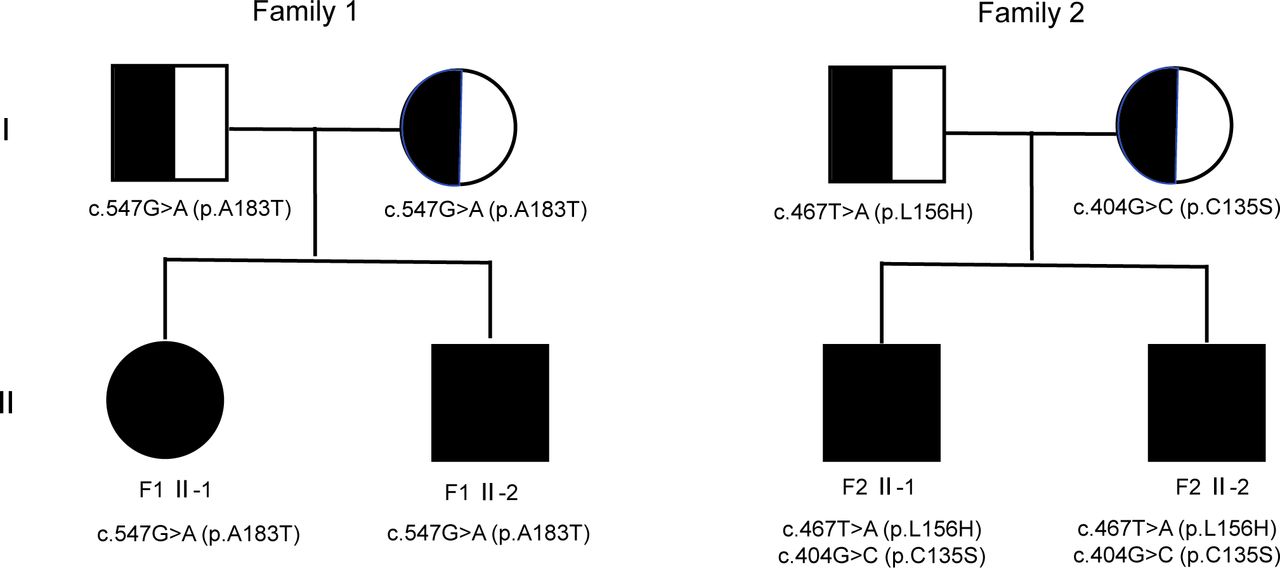
Pedigree of the families. Black/white represents the healthy parents with heterozygous variant in NDUFV2, while the black symbols depict the affected patients with homozygous or compound heterozygous variant in NDUFV2.
Next-generation sequencing
Targeted next-generation sequencing was performed for each patient, which contained 115 genes involved in vanishing white matter disease, Krabbe disease, adrenoleukodystrophy, metachromatic leukodystrophy, Alexander disease and so on. No suspected disease-causing mutations were found. Therefore, we further applied whole-genome sequencing (WGS) or whole-exome sequencing (WES) to the individuals’ DNA extracted from peripheral blood. Family 1 (including the two sibling patients and their parents) was investigated by WGS, and Family 2 was investigated by WES. WES coding regions were enriched using the Agilent SureSelect Human All Exon V6 Kit followed by sequencing as 150 bp paired-end runs on an Illumina HiSeq2000. WGS sequencing libraries were prepared using Illumina TruSeq Nano Kits and sequenced on Illumina HiSeq X sequencers via 2×150 bp reads. Reads were aligned to the human reference genome (UCSC Genome Browser build hg19, https://genome.ucsc.edu/) using Burrows-Wheeler Aligner20 (BWA, http://bio-bwa.sourceforge.net/). ANNOVAR21 (http://annovar.openbioinformatics.org/) software was used for variant filtering and annotation. Rare variants were subsequently filtered by a minor allele frequency (MAF) lower than 0.1% in 1000 Genomes Project22 (http://www.internationalgenome.org/), Exome Aggregation Consortium23 (ExAC, http://exac.broadinstitute.org/) and gnomAD24 (https://gnomad.broadinstitute.org/) databases. The missense variants in highly conserved amino acid residues predicted by PolyPhen-225 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster26 (http://www.mutationtaster.org/), SIFT27 (https://sift.bii.a-star.edu.sg/) and M-CAP28 (http://bejerano.stanford.edu/mcap/) tools, and those causing protein truncation or frameshift were considered as pathogenic. Furthermore, the potential pathogenic variants should also be shared by the two patients of the same family. Variants were interpreted according to the guidelines published by ACMG using the HGVS (https://varnomen.hgvs.org/) nomenclature.29 Sanger sequencing was used to confirm the identified mutations.
Muscle histopathology study
Muscle biopsy was carried out for F1 II-1. Muscle samples used for histopathological study were frozen after collection in liquid N2-cooled isopentane and stored at −80°C until use. The samples were assessed for H&E, modified Gomori trichrome, succinate dehydrogenase (SDH), NADH-tetrazolium reductase (NADH-TR), cytochrome oxidase (COX), glycogen and lipid staining.30
Mitochondrial respiratory chain (MRC) enzyme assay
Skin biopsy was performed for II-1 and II-2 of Family 1. The skeletal muscle used for the MRC enzyme assay was stored at −80°C before use. Activities of MRC complexes I, II, II+III, III, IV and the mitochondrial marker enzyme citrate synthase (CS) were assayed in the crude post-600 g supernatant of the muscle sample and in isolated mitochondria from skin fibroblasts using spectrophotometric assays as described previously.12 31 32 The enzyme activities of each complex are presented as the percentage of the normal control mean relative to appropriate reference enzyme activities, such as CS or MRC complex II. Enzyme activity was defined as being decreased at a level of <40% in a cell line or <30% in tissue.33
Oxygen consumption rate (OCR) measurement
OCR was measured in fibroblasts from the two patients (F1 II-1 and II-2) with a XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, Massachusetts, USA). Samples were prepared as previously described.12 34 In each run, we measured two controls along with the patient samples. The control cell lines used were derived from fHDF (Toyobo Biotech Support Department, Japan). Each control and patient fibroblast cell line was seeded in at least 14 wells of two XF96 cell culture microplates (Seahorse Bioscience) at a density of 20 000 cells/well in 80 µL of growth medium and incubated at 5% CO2 and 37°C overnight. The following day, the growth medium was replaced with 160 µL of 25 mM glucose medium or 10 mM galactose medium, respectively, and the cell culture microplate was placed into a CO2-free incubator at 37°C for 60 min before measurement. After measurement of the basal OCR, 10 µM oligomycin, 4 µM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and 20 µM rotenone were added sequentially, and the OCR was recorded after each addition.34 The maximum respiration rate (MRR) corresponds to the OCR after FCCP injection minus the rotenone-insensitive OCR. MRR was expressed as a percentage relative to the average of controls, in which a reduction to <71.6% was considered to represent a significant decline.12
Complementation of NDUFV2 mutation by cDNA expression
The fibroblasts of F1 II-1 and II-2 were obtained by skin biopsy. The complementation assay was based on the widely used ViraPower HiPerform Lentiviral Expression System. The wild-type (wt) cDNAs from NDUFV2 were cloned into the pLenti6.3 vector, and virions were obtained as previously described.34 Patient and control fibroblasts from healthy individuals were infected with viral supernatant and selected on exposure to 5 µg/mL blasticidin. The OCR of infected fibroblasts was measured, and the rescue effect was analysed.
Results
Patients' history and clinical presentations
Family 1
F1 II-1, the first child of healthy, non-consanguineous parents from different countries, had normal prenatal and perinatal histories, with a birth weight of 3.15 kg. Her parents cannot accurately describe her developmental milestones, but she could sit, stand alone and speak ‘baba and mama’ when she was 1 year old. At the age of 14 months, she suddenly showed signs of developmental regression after a fever. Gradually, she was not able to stand or sit alone, nor she could speak; she also showed difficulties opening her mouth and eating. MRI showed a characteristic pattern of cystic changes in the corpus callosum and deep periventricular white matter (figure 2A–F). After IVIG, ribavirin and oxiracetam treatment, her clinical symptoms gradually improved. At the age of 2 years, she could again walk alone. MRI seemed relatively stable up to 6 years and 3 months old (figure 2H–M). When examined at 7 years and 2 months old, her neurological condition was stable, with slight infantile behaviour. Although her speech and gait were normal, her school examination scores were poor. Physical examination revealed that her height and weight were both below the third centile (17 kg, 112 cm), intermittent external strabismus could be observed in her left eye and bilateral knee tendon reflexes were active. After genetic diagnosis, she was treated with thiamine (150 mg/day), riboflavin (150 mg/day), coenzyme Q10 (150 mg/day), L-carnitine (1000 mg/day) and vitamins C (100 mg/day) and E (100 mg/day) for only 2–3 months. Up to the age of 7 years and 8 months, she walked, ran and jumped normally and had relatively normal cognitive abilities.

Brain MRI of F1 II-1 (A) at the age of 16 months (A–F) and 6 years and 3 months (H–M), and F1 II-2 (B) at the age of 12 months (O–T) and 4 years and 8 months (U–Z). A, B, H, I, Q, R, W and X—axial T1-weighted images; C, D, J and K—axial T2-weighted images; E, F, O, P, U and V—sagittal T1-weighted images and L, M, S, T, Y and Z—axial T2-FLAIR images. MRI of F1 II-1 showed long T1 and T2 signals with some cystic changes in the corpus callosum and cerebral hemispheric white matter, which were relatively stable up to 6 years and 3 months of age. Earlier MRI of F1 II-2 showed long T1 and T2 signals with some cystic changes in cerebral hemispheric white matter, mainly involving periventricular white matter and centrum semiovale, with sparing of the corpus callosum, while the later MRI showed worsening and widening of cystic lesions involving the white matter and the corpus callosum.
F1 II-2, the second child of Family 1, was born after an uneventful perinatal period. He started to present mild developmental delay at the age of 12 months when he was able to sit alone but could not climb or stand. After that, he suddenly displayed developmental regression with no obvious predisposing causes. MRI showed long T1 and T2 signals with some cystic changes in cerebral hemispheric white matter, mainly involving periventricular white matter and centrum semiovale, with sparing of the corpus callosum (figure 2O–T). The symptoms then abated, and he remained stable for some time. At the age of 20 months, he could walk alone. However, when he was 4 years and 8 months old, he had episodes of neurological deterioration, which were characterised by irritability, salivation, dysarthria, dysphagia, dystonia, developmental regression, strabismus and pyramidal tract impairment. During the most serious episode, he could only speak word by word with an unclear voice, and he could neither hold his head up nor sit. MRI showed diffuse cystic changes involving the cerebral hemispheric white matter and corpus callosum (figure 2U–Z), which was worse than before. Cerebral spinal fluid (CSF) lactate was 2.22 mmol/L (normal <2.8 mmol/L), and CSF cell count, protein, glucose, chloride and cultures were normal. Urine organic acid and plasma amino acid results were normal. After oxiracetam, thiamine and mecobalamin treatment, his movement and language gradually improved. When examined at 5 years and 8 months old, his condition was stable, with a slightly unstable gait and low intelligence but normal speech. Physical examination revealed external strabismus in his left eye as well as a spastic gait, accompanied by hypertonia, bilateral active knee tendon reflexes and positive Babinski signs. His weight was at the 10th centile (17 kg), and his height was between the 10th and 25th centiles (112 cm). After the genetic diagnosis was established, he received thiamine (150 mg/day), riboflavin (150 mg/day), coenzyme Q10 (150 mg/day), L-carnitine (1000 mg/day) and vitamins C (100 mg/day) and E (100 mg/day) for 3 months. Up to the age of 6 years and 2 months, he walked, ran and jumped with a slightly abnormal gait but had relatively preserved cognitive abilities.
Family 2
F2 II-1, the first child born of healthy, non-consanguineous parents, was born after an uneventful perinatal period. Before 4 months of age, his developmental milestones were normal, but after that, his development was arrested. When he was admitted to the hospital at the age of 11 months, he had developmental delay but relatively preserved cognitive function, which showed that he could turn over and understand simple instructions but could not sit nor stand alone. Physical examination indicated hypertonia of both lower extremities, bilateral active knee tendon reflexes and positive Babinski signs. Serum lactate, urine organic acid and plasma amino acid levels were normal. MRI showed a characteristic pattern of cystic changes in the deep periventricular white matter. He was clinically diagnosed with leukoencephalopathy and treated with coenzyme Q10 (30 mg/day), L-carnitine (500 mg/day), thiamine (20 mg/day), riboflavin (10 mg/day), vitamin B6 (20 mg/day) and mecobalamin (0.5 mg/day) for only 1 year. During the clinical course, he presented mild development regression after an infection but gradually returned to baseline with the recovery from the infection. Followed up to 8 years and 6 months old, he could turn over and sit alone but could not stand or walk, yet his cognitive function was relatively preserved.
F2 II-2, the second child of Family 2, had normal prenatal and perinatal histories and normal developmental milestones before the age of 1. He had gradual developmental regression after vaccine inoculation at the age of 1 year. When he was admitted to the hospital at the age of 13 months, he could not turn over nor stand, but his cognitive function was relatively preserved. Physical examination showed hypertonia of both lower extremities, bilateral active knee tendon reflexes and positive Babinski signs. Serum lactate, urine organic acid and plasma amino acid levels were normal. MRI showed a characteristic pattern of cystic changes in the deep periventricular white matter. He was clinically diagnosed with leukoencephalopathy and treated like his brother with coenzyme Q10 (30 mg/day), L-carnitine (500 mg/day), thiamine (20 mg/day), riboflavin (10 mg/day), vitamin B6 (20 mg/day) and mecobalamin (0.5 mg/day). Up to the age of 4 years and 11 months, he walked, ran and jumped with a slightly unstable gait but had relatively preserved cognitive abilities.
Next-generation sequencing (WGS and WES)
We selected the potential causative variants by excluding those silent (noncoding or synonymous), common (MAF >0.1%) or not following autosomal-recessive inheritance. Those variants have been provided in the online supplemental tables 1 and 2. Furthermore, the missense variants in highly conserved amino acid residues predicted by PolyPhen-2, MutationTaster, SIFT and M-CAP tools, and those causing protein truncation or frameshift were considered as pathogenic. Ultimately, only NDUFV2 has been identified as the pathogenic gene in the two families.
Supplemental material
Supplemental material
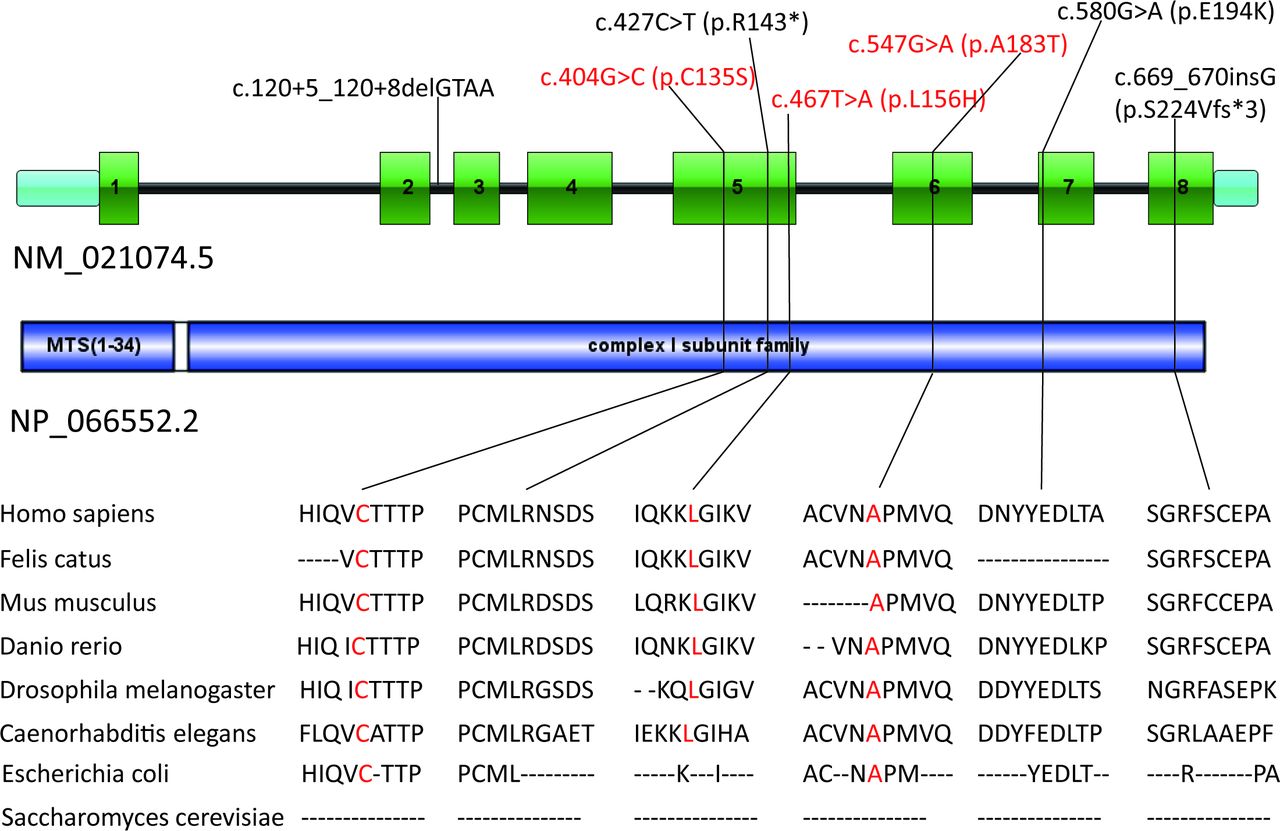
The WGS performed in the affected individuals and their parents of Family 1 revealed a homozygous missense variant c.547G>A (p.A183T) in NDUFV2 (NM_021074.5) in the affected individuals and the heterozygous missense variant in NDUFV2 in their parents encoding a mitochondrial Co I subunit (online supplemental table 1). This variant is a novel mutation that has not been reported before. The WES performed in the affected individuals of Family 2 revealed compound heterozygous missense variants c.467T>A (p.L156H) and c.404G>C (p.C135S) in the same gene, NDUFV2 (online supplemental table 2). The three variants are all absent in the 1000 Genomes Project, Exome Aggregation Consortium (ExAC) and gnomAD databases and are all predicted as deleterious by a group of tools, including SIFT, Polyphen-2, MutationTaster and M-CAP. Sequence alignments of the NDUFV2 proteins from bacteria to humans showed that all the affected amino acids are highly conserved (figure 3).

NDUFV2 gene structure shows the localisation of the reported and investigated (red) gene variations. Conservation of the affected amino acid residues is presented in the alignment of homologs across different species.
Muscle histopathology study and MRC enzyme assay
There were no abnormalities detected using H&E, Gomori trichrome, SDH, NADH-TR, COX, NSE, glycogen and lipid staining, and no ragged red, SDH-rich, ragged blue or COX-negative fibres were detected. Low activity of Co Ⅰ was found in both affected individuals’ fibroblasts and in the muscle of II-1 of Family 1. Co Ⅰ activity in F1 II-2 was lower than that in F1 II-1 (table 1).
Mitochondrial respiratory chain assay from fibroblasts and muscle
OCR and complementation studies in fibroblasts
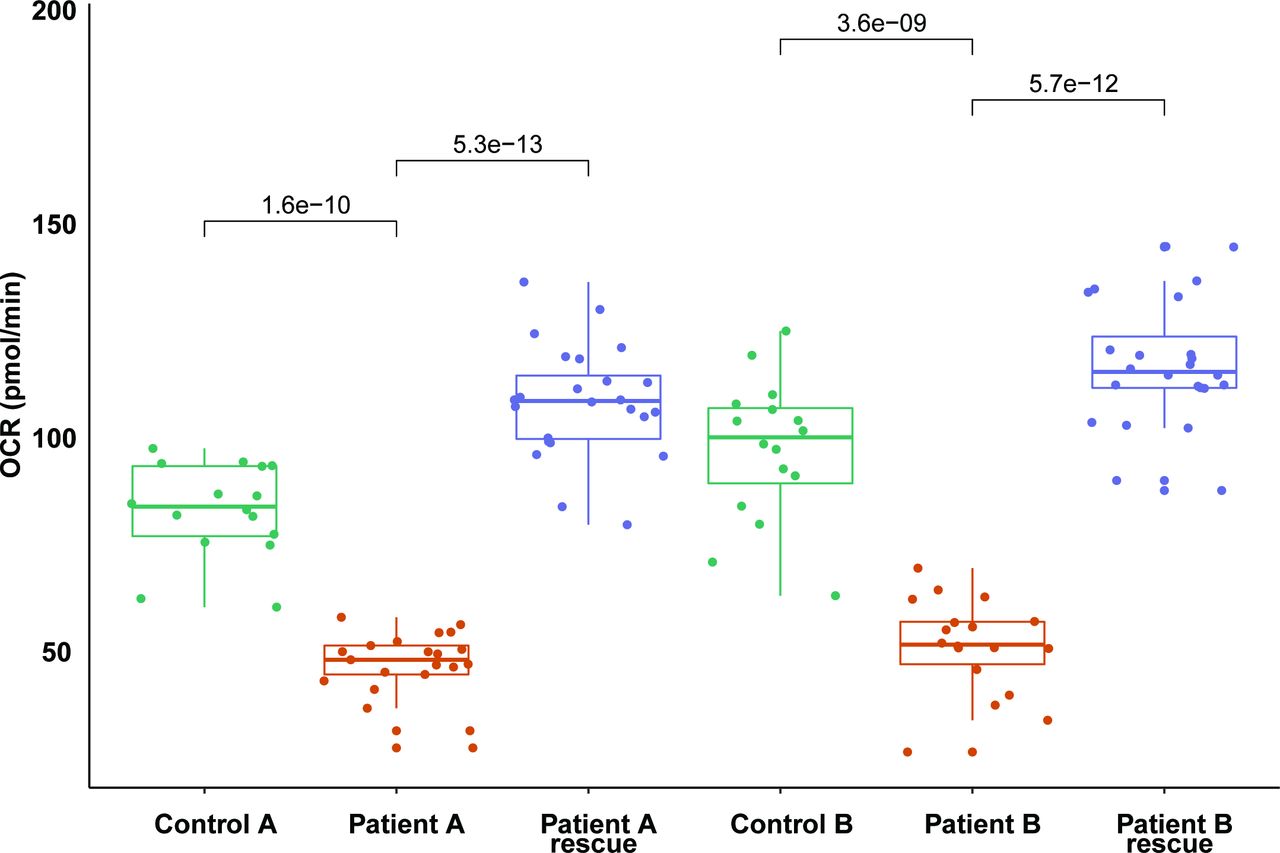
In addition to measuring the OCR in cells grown in glucose medium, we measured the OCR in cells grown in galactose medium, a condition that forces cells to depend on mitochondrial respiration rather than glycolysis for ATP production. A significant decline of MRR with glucose and galactose medium was found in both siblings of Family 1 compared with the control. The OCR of F1 II-1 was 64% and 53% relative to the average of controls with glucose and galactose medium, respectively, while the OCR of F1 II-2 was 56% and 43%, respectively. Next, we took advantage of the cellular phenotype in a complementation experiment. The OCR of both Family 1 patients increased to normal values after transduction with a recombinant lentiviral construct expressing the wt cDNA of NDUFV2 (figure 4), which validates the pathogenic role of the novel NDUFV2 variant in defective mitochondrial respiration.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Complementation of NDUFV2 variants by cDNA expression. Individual OCR expressed as pmol O2/min/cell in cell lines of the two patients of family 1 with NDUFV2 mutations compared with controls and complemented patient cell lines. Significant rescue effect (p<0.01) can be found. OCR, oxygen consumption rate.
Discussion
We report sibling pairs from two Chinese families presenting a similar clinical course of recurring episodes of acute or subacute developmental regression that appeared in the first year of life (table 2). This decline was followed by gradual remissions and prolonged periods of stability. These patients had similar MRI findings that showed leukoencephalopathy with multiple cavities. Gene sequencing revealed that all four patients carried pathogenic novel variants in the NDUFV2 gene.
Clinical manifestation summary of the four patients from two Chinese families
Since the first disease gene, NDUFS1, was reported to be related to PCL in 2011,5 NDUFV1 and other genes related to mitochondrial function and energy metabolism have been reported, while mutations in NDUFS1 and NDUFV1 seem to be more common.6–8 NDUFV1, NDUFV2 and NDUFS1-encoding subunits compose the N module of Co I, which oxidises NADH.6 11 With a similar function, NDUFV2 was supposed to be another causal gene of PCL. NDUFV2 is highly expressed in the brain, mainly in neurons, astrocytes and oligodendrocytes, so a NDUFV2 mutation can induce dysfunction of neurons, astrocytes and oligodendrocytes, causing the corresponding clinical syndromes. To date, six mutations have been described in NDUFV2, most of which result in Leigh syndrome, but none has been reported to be associated with PCL. In this study, we identified three novel NDUFV2 mutations in patients with PCL. There are several lines of evidence suggesting the pathogenicity of the following NM_021074.5 mutations: c.547G>A (p.A183T), c.467T>A (p.L156H) and c.404G>C (p.C135S) in NDUFV2.29 First, homozygous and compound heterozygous mutations occurred in sibling pairs of two families with similar clinical and neuroimaging phenotypes, segregating from the two heterozygous healthy parents. Second, the three mutations were all absent from controls in the 1000 Genomes Project, ExAC and gnomAD databases, were all in highly conserved amino acid residues and were all predicted as deleterious by a group of tools, including SIFT, Polyphen-2, MutationTaster and M-CAP. Third, the MRC enzyme assay showed Co I deficiency in the fibroblasts of both affected individuals and in the muscle of the elder sister of Family 1. Finally, the two patients of Family 1 showed a significant decrease in MRR compared with the control, and a significant rescue effect could be shown by functional complementation of patient-derived fibroblast cell lines, which is an established method to validate the disease-causing variant in various genes.34
In the present patients, CNS involvement is the main clinical presentation. Common clinical features include developmental regression, followed by gradual remissions and prolonged periods of stability. Brain MRI revealed abnormalities in the white matter of all four patients. Our results suggest that leukoencephalopathy in these patients is caused by NDUFV2 mutations. The clinical features described in these patients differ from the previously described NDUFV2-associated neurological diseases.
Data availability statement
Data are available on reasonable request. The data that support the findings of this study are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The Ethics Committee of each hospital approved the study, and prior informed consent was obtained from the patients’ parents.
Acknowledgments
The authors would like to thank the families for their participation in this study. We thank AJE Team (www.aje.com) for editing a draft of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ZL and LZ contributed equally.
Contributors ZL collected and analysed the data and was responsible for drafting the manuscript. LZ analysed the genetic data and was responsible for drafting the manuscript. CR, MX, SL, RB, YW, LC and SS contributed to phenotyping and to acquiring and analysing the clinical and histopathological data. ME and HP conducted functional experiments, collected and analysed the functional data and contributed to the manuscript. ZL, MS, MO-T and KM conducted the enzyme assay and OCR experiments and collected and analysed these data. TS, HP and FF were responsible for conceiving, designing and supervising the study and for writing the manuscript. All authors revised the manuscript for intellectual content.
Funding This study was funded by the Capital Health Development Research Foundation Project (2018-2-2096), the National Natural Science Foundation of China (81541115) and the Henan Provincial Key Laboratory of Children’s Genetics and Metabolic Diseases Foundation (SS201904).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.