Article Text

Abstract
Background Pathogenic heterozygous SIX1 variants (predominantly missense) occur in branchio-otic syndrome (BOS), but an association with craniosynostosis has not been reported.
Methods We investigated probands with craniosynostosis of unknown cause using whole exome/genome (n=628) or RNA (n=386) sequencing, and performed targeted resequencing of SIX1 in 615 additional patients. Expression of SIX1 protein in embryonic cranial sutures was examined in the Six1 nLacZ/+ reporter mouse.
Results From 1629 unrelated cases with craniosynostosis we identified seven different SIX1 variants (three missense, including two de novo mutations, and four nonsense, one of which was also present in an affected twin). Compared with population data, enrichment of SIX1 loss-of-function variants was highly significant (p=0.00003). All individuals with craniosynostosis had sagittal suture fusion; additionally four had bilambdoid synostosis. Associated BOS features were often attenuated; some carrier relatives appeared non-penetrant. SIX1 is expressed in a layer basal to the calvaria, likely corresponding to the dura mater, and in the mid-sagittal mesenchyme.
Conclusion Craniosynostosis is associated with heterozygous SIX1 variants, with possible enrichment of loss-of-function variants compared with classical BOS. We recommend screening of SIX1 in craniosynostosis, particularly when sagittal±lambdoid synostosis and/or any BOS phenotypes are present. These findings highlight the role of SIX1 in cranial suture homeostasis.
- musculoskeletal diseases
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
The identification of disease-causing mutations has greatly benefited from the introduction of next-generation sequencing technologies. In particular, whole exome and genome sequencing (WES/WGS) have both accelerated the discovery of new genes involved in rare genetic diseases and, because of their unbiased nature, have enabled expansion of clinical phenotypes associated with known disease genes. Moreover retrospective clinical investigation of the patient and family members can reveal additional, previously unrecognised clinical features.1
The initial aim of this work was to investigate a possible genetic cause in a proband affected by speech and language delay, sensorineural hearing loss (HL) and craniosynostosis (CRS), the premature fusion of one or more cranial sutures of the skull. We identified a de novo mutation in SIX1, which encodes a homeodomain-containing transcription factor of the sine oculis class, originally described in Drosophila.2 Further analysis of additional unsolved CRS cases using WES/WGS, RNA sequencing or targeted resequencing identified heterozygous variants in SIX1 in seven further patients with CRS from six unrelated families. Dominantly inherited SIX1 variants were previously reported in branchio-otic syndrome (BOS; MIM 608389),3 4 non-syndromic HL (MIM 605192)5 and (rarely) in branchio-oto-renal (BOR) syndrome, associated in addition with renal malformation.3 This work uncovers a previously unrecognised role of SIX1 in the maintenance of cranial suture patency.
Subjects and methods
Patients
Written informed consent was obtained from all participants/legal guardians. The clinical diagnosis of CRS was confirmed by three-dimensional CT scanning of the skull. When clinically indicated, samples were tested for mutation hotspots in FGFR2, FGFR3, TWIST1, TCF12 and ERF, and significant chromosome aneuploidy was investigated using array comparative genomic hybridisation; samples harbouring mutations known to cause CRS were excluded.
Genetic analyses
WES or WGS of unrelated probands with CRS of unknown cause (n=103 and n=525 in Oxford and Yale cohorts, respectively) and subsequent bioinformatic analyses were performed as previously described.6 7 RNA sequencing of patient osteoblast cell lines (n=386) was previously described.8 Targeted screening for SIX1 variants was performed by resequencing of multiplexed PCR products encompassing the coding regions and intron/exon boundaries of SIX1 (NM_005982, ENST00000247182) in an additional 615 unsolved patients with CRS (mutation negative for the major known causes). Primer sequences are provided in online supplemental table S1 and the clinical diagnoses of probands screened in online supplemental table S2. Variant calls and coverage information were obtained using amplimap.9 Validation and segregation analysis of variants were undertaken by dideoxy-sequencing of PCR products from genomic DNA. Correct sample relationships were confirmed either by comparison of trio-WES data or by analysis of 13 microsatellite markers located on different chromosomes. Previously reported SIX1 variants were sourced from HGMD Professional V.2019.4, available online communications to the European Society of Human Genetics, and PubMed searches.
Supplemental material
Six1-LacZ reporter analysis
Heads from heterozygous E18.5 Six1nLacZ (Six1tm1Mair ) mice10 were fixed for 30 min in 4% paraformaldehyde at room temperature and incubated in 15% sucrose overnight at 4°C before embedding in Optimal Cutting Temperature (OCT) compound and freezing in isopentane cooled with liquid nitrogen. One in every 20 frontal sections (10 µm thick) was harvested, incubated in X-gal solution for 2.5 hours at 37°C, mounted on glycerol and imaged with 4× and 10× objectives of an upright fluorescent microscope (Olympus BX63), equipped with an ORCA-Flash4.0 LT Hamamatsu camera, using Metamorph V.7 software.
Results
Identification of SIX1 variants in CRS
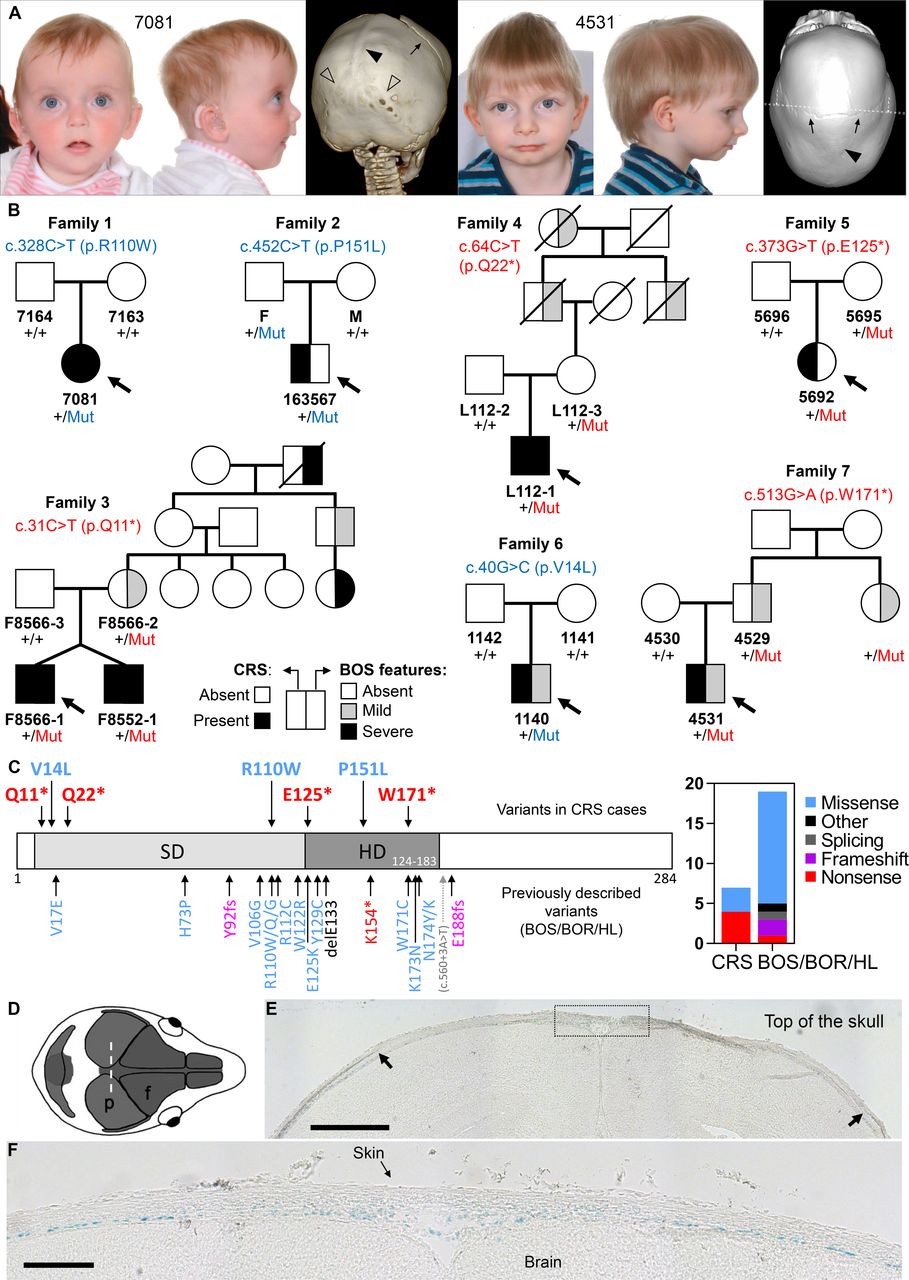
Parent–child trio-based WES was performed on proband 7081, affected by sagittal and bilambdoid CRS (figure 1A, left), bilateral sensorineural HL, and speech and language delay. This identified a de novo SIX1 c.328C>T (p.R110W) mutation (family 1 in figure 1B, table 1). SIX1 variants, including p.R110W, were previously reported in individuals with BOS/BOR syndromes (dominantly inherited disorders characterised by variable hearing impairment, preauricular pits, branchial defects±kidney malformations) or non-syndromic HL (figure 1C).3–5 No additional candidate variants were identified to explain the CRS in proband 7081, who retrospectively was noted to manifest additional features of BOS, including bilateral preauricular skin tags, ear pits and a possible sinus over the right sternocleidomastoid (see online supplemental case report).
{kind=link}
(A) Clinical photographs and CT head scans of individuals 7081 aged 7 months (left) and 4531 aged 2.5 years (right), probands of families 1 and 7, respectively. Note the fused sagittal sutures (filled arrowheads), and in 7081 lambdoid sutures (unfilled arrowheads). The coronal sutures are patent (arrows). (B) Pedigrees of families harbouring missense (blue lettering) or nonsense (red lettering) variants in SIX1; individuals affected with CRS shown with black fill (left side of the pedigree symbol) and individuals with BOS features shown with black or grey fill according to severity (right side of the pedigree symbol; mild: preauricular pit or late-onset HL; severe: branchial cyst/fistula or congenital HL). Genotypes are indicated for all available samples. (C) SIX1 variants in congenital disease. (Left) Domain structure of the human SIX1 protein showing the locations of the six domain (SD) and homeodomain (HD). Pathogenic variants, colour-coded according to the key at far right, are shown (above the protein cartoon) for the seven CRS-associated variants identified in this work and (below the cartoon) for previously identified variants in BOS/BOR/HL syndromes. (Right) The chart represents the number of different variants of each pathogenic class identified in CRS or in BOS/BOR/HL. (D–E) SIX1 protein expression revealed by X-gal staining in frontal sections (10 µm thick) of E18.5 Six1nLacZ/+ mouse heads. (D) Representation of the mouse skull from above to illustrate the plane of section (coronal) in the histological sections shown (p, parietal bone; f, frontal bone). (E) Arrows indicate the superior margins of the parietal bones, and the dotted box (enlarged in a nearby section in (F) shows the sagittal suture. Blue nuclei (X-gal positive) indicate SIX1 expression. Scale bars: 500 µm (E) and 150 µm (F). BOR, branchio-oto-renal; BOS, branchio-otic syndrome; CRS, craniosynostosis; HL, hearing loss.
SIX1 variants identified in subjects with CRS
To determine whether CRS was causally associated with the SIX1 mutation, we interrogated existing cohorts of genetically undiagnosed CRS investigated by either WES/WGS (n=628)6 7 or RNA sequencing (n=386).8 This highlighted four additional families carrying rare heterozygous SIX1 variants (families 2–5; figure 1B, table 1). No additional candidate variants explained the CRS in these cases. To investigate further the significance of SIX1 variants, we performed targeted resequencing of SIX1 in a cohort of 615 additional patients with any type of CRS, leading to the discovery of two further families with heterozygous SIX1 variants (families 6 and 7; figure 1B, table 1). In addition to the initial finding (family 1), one of the variants from the cohort screen (c.40G>C; p.V14L) arose de novo in the proband (family 6), whereas in the remaining five families (families 2–5 and 7) the variant had been transmitted from one of the parents. Dideoxy-sequence verification of all SIX1 variants is shown in online supplemental figure S1. Of 1629 probands analysed, SIX1 variants were present in 3 of 822 (0.4%) sagittal synostosis, 4 of 112 (3.6%) multisuture (excluding bicoronal or bilambdoid) synostosis and 0 of 695 with other types of suture fusion (online supplemental table S2).
Phenotype and genotype–phenotype correlation
Overall we identified eight patients with CRS heterozygous for variants in SIX1, from seven unrelated families. The variants comprised de novo missense (n=2) or inherited nonsense (n=4) or missense (n=1) changes (figure 1B, table 1). The missense changes are located at residues conserved in all six human SIX paralogues, as well as Drosophila sine oculis (online supplemental box S1); Combined Annotation Dependent Depletion (CADD) scores are correspondingly high (24.4–33). Interestingly, all individuals with CRS had sagittal synostosis (figure 1A), in five instances combined with fusion of both coronal (n=1) or both lambdoid (n=4) sutures (table 1). Six of these eight individuals (75%) presented additional clinical features compatible with BOS (detailed descriptions in the online supplemental case reports), notably branchial arch defects (preauricular pits or tags, neck swellings or sinuses, n=5) and/or HL (n=2). No symptomatic renal abnormalities were documented, consistent with a low incidence of renal disease caused by SIX1 variants in previous reports4; however targeted renal imaging was undertaken in only three patients. In three of the inherited cases, retrospective analysis indicated the presence of BOS-related features in additional family members and apparent non-penetrant carrier status in others (figure 1B, table 1).
Considering the predicted functional consequences of SIX1 variants, four of the seven different variants (57%) are stop-gain, representing a 2.7-fold enrichment of loss-of-function (LoF) variants in CRS compared with the spectrum previously reported in BOS/BOR/HL, which are more often caused by missense variants (73.7% of the 19 different pathogenic variants identified in 27 unrelated families reported in the literature; figure 1C, online supplemental table S3). Although this difference is not significant (p=0.15, two-tailed Fisher’s exact test), there is apparent enrichment (p=0.01) when only the nonsense variants are considered (4 of 7 in CRS vs 1 of 19 in BOS/BOR/HL).
SIX1 expression in the sagittal suture
To investigate whether SIX1 is expressed in cranial sutures, we examined a previously reported transgenic mouse line with the nls-LacZ reporter inserted into the first exon of Six1.10 X-gal staining of frontal sections of E18.5 Six1nLacZ/+ embryonic heads demonstrated β-galactosidase activity in a layer basal to the growing bones, likely corresponding to the dura mater, and extending into the mesenchyme of the future sagittal suture (figure 1D–F). By contrast, no significant staining was observed in the osteogenic fronts or mid-sutural mesenchyme of the coronal sutures (online supplemental figure S2).
Discussion
This study identifies enrichment of heterozygous deleterious SIX1 variants in patients with CRS, expands the phenotype of SIX1-related disorders and provides evidence for a previously unrecognised role of SIX1 in normal homeostasis of the cranial sutures.
Of note, sagittal suture fusion was present in all eight affected individuals, an unusual pattern as monogenic types of CRS more commonly involve the coronal sutures.11 In four subjects, the sagittal synostosis was present together with the involvement of both lambdoid sutures (‘Mercedes-Benz’ pattern), a rare combination of suture fusions present in 23 of the 1629 probands screened for SIX1 mutations (online supplemental table S2). In a cohort of 4250 unselected CRS cases treated in a single department, only 39 patients were diagnosed with combined sagittal and bilambdoid synostosis12; our finding that 4 of 8 patients with SIX1 pathogenic variants exhibited the Mercedes-Benz pattern represents a significant enrichment (p<10−6, two-tailed Fisher’s exact test).
In addition to CRS, six of the eight patients exhibited additional clinical features compatible with SIX1-related BOS/BOR/HL. However, the associated anomalies were sometimes minor (such as ear pits) and were retrospectively identified in some cases. Significant HL was documented in only two of eight individuals with CRS. Whereas de novo missense mutations accounted for two sporadic cases, parental transmission occurred in the other five families, and in three of these an extended family history compatible with BOS±isolated HL was evident (figure 1B). Although intrafamilial phenotypic variability is frequent in SIX1-related disease, non-penetrance is uncommon3 4; by contrast, non-penetrance occurred in all five families showing parental transmission of the variant, including three confirmed SIX1-heterozygous individuals (figure 1B, families 2, 4 and 5). Notably, in four of these families, the SIX1 variant encodes a nonsense change, only previously described once in BOS/BOR/HL, in a patient additionally noted to have macrocephaly.13
In summary, whereas the classic BOS/BOR/HL-associated variants are usually missense substitutions that may have dominant-negative activity (figure 1C),4 14 we propose that heterozygous SIX1 LoF variants are associated with a haploinsufficiency phenotype that overlaps with BOS/BOR/HL, but includes a propensity to CRS in some individuals and non-penetrance in others. According to the gnomAD (V.2.1.1) database,15 SIX1 is moderately constrained (probability of LoF intolerance(pLI)=0.65; observed/expected=0.17 (0.07–0.52) for LoF alleles), with 10 LoF alleles observed in a minimum of 238 186 alleles surveyed. Although this supports that halving the effective SIX1 dosage sometimes has mild consequences, the enrichment of CRS associated with SIX1 LoF alleles (4 of 3258) is highly significant (p=0.00003, Fisher’s exact test).
SIX1 is a homeodomain-containing transcription factor that is essential for normal development of several organs. In the head, Six1 is expressed in different lineages in avian and murine embryos, and Six1-deficient mice display severe craniofacial malformations during embryonic development.16 17 To our knowledge, however, Six1 expression has not been formally analysed in the cranial sutures. Considering the specificity of the sagittal suture involvement in all the SIX1-positive cases, we focused our analysis on this suture and used a nls-LacZ reporter gene (inserted at the Six1 locus)10 to mirror the spatiotemporal pattern of SIX1 expression in heterozygous mouse embryos (E18.5). The presence of SIX1 associated with a basal layer between the growing bone and the brain is intriguing. This layer most likely represents the dura mater, which was recently shown in a single cell transcriptomic analysis to be enriched for Six1 expression.18 Paracrine signals from the dura mater contribute to maintenance of suture patency and closure,19 by coordinating availability of secreted factors such as fibroblast growth factors and β-type transforming growth factors, signalling pathways potentially regulated by SIX1.20 However, to our knowledge, no genetic mutants have been shown to act primarily by disturbing the dura mater–suture interactions. This work therefore provides a starting point to investigate the contribution of SIX1 dosage to suture homeostasis and the downstream targets perturbed in the presence of pathogenic variants.
In summary, both missense and nonsense variants in SIX1 confer a substantially increased risk of CRS. We recommend testing for SIX1 variants in undiagnosed CRS with sagittal involvement, especially when occurring in combination with lambdoid synostosis and/or associated with BOS/BOR/HL-related clinical features in the proband or extended family.
Ethics statements
Patient consent for publication
Ethics approval
The clinical and genetic studies were approved by respective medical ethics boards (UK: London-Riverside Research Ethics Committee, reference 09/H0706/20; Brazil, Instituto Biociencias, University of São Paulo Ethics Committee in Human Research, reference #050/2006; USA: Yale Human Investigation Committee Institutional Review Board and Seattle Children’s Hospital Institutional Review Board, reference 12394). Animal experiments were carried out in accordance with the European STE 123 and the French national charter on the Ethics of Animal Experimentation. Protocols were approved by the Ethical Committee of Animal Experiments of the Institut Cochin, CNRS UMR 8104, INSERM U1016, and by the Ministère de l’éducation nationale, de l’enseignement et de la recherche, n° APAFIS#15 699-2018021516569195.
Acknowledgments
We thank all the families for their participation in this study. We thank the staff at the MRC-WIMM facility for DNA sequencing.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice The article has been corrected since it was published Online First. The missing abstract has been added to the article.
Contributors DRB, MLC, DJ, JEVM, ATT, SAW and AOMW undertook patient recruitment. EC, RA, JAG, SJM, ATT and SRFT performed the genetic analyses, supervised by MLC, MRP-B, RPL and AOMW. MW and PM analysed the mouse reporter line. EC and SJM performed the bioinformatics analyses. EC, PM and AOMW drafted the manuscript, with clinical input from DRB, MLC, JEVM and ATT. All authors approved the final version.
Funding This work was supported by FAPESP/CEPID (2013/08028-1 and 303712/2016-3 to DRB and MRP-B), AFM (17406 to PM), NIH/NIDCR (5R01DE018227-10 to MLC), the NIHR Oxford Biomedical Research Centre (AOMW) and the NIHR UK Rare Genetic Disease Research Consortium, the MRC through the WIMM Strategic Alliance (G0902418 and MC UU 12025), the VTCT Foundation (SRFT and AOMW) and Wellcome (102731 to AOMW).
Disclaimer The views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.