Article Text

Abstract
SOX10 belongs to a family of 20 SRY (sex-determining region Y)-related high mobility group box-containing (SOX) proteins, most of which contribute to cell type specification and differentiation of various lineages. The first clue that SOX10 is essential for development, especially in the neural crest, came with the discovery that heterozygous mutations occurring within and around SOX10 cause Waardenburg syndrome type 4. Since then, heterozygous mutations have been reported in Waardenburg syndrome type 2 (Waardenburg syndrome type without Hirschsprung disease), PCWH or PCW (peripheral demyelinating neuropathy, central dysmyelination, Waardenburg syndrome, with or without Hirschsprung disease), intestinal manifestations beyond Hirschsprung (ie, chronic intestinal pseudo-obstruction), Kallmann syndrome and cancer. All of these diseases are consistent with the regulatory role of SOX10 in various neural crest derivatives (melanocytes, the enteric nervous system, Schwann cells and olfactory ensheathing cells) and extraneural crest tissues (inner ear, oligodendrocytes). The recent evolution of medical practice in constitutional genetics has led to the identification of SOX10 variants in atypical contexts, such as isolated hearing loss or neurodevelopmental disorders, making them more difficult to classify in the absence of both a typical phenotype and specific expertise. Here, we report novel mutations and review those that have already been published and their functional consequences, along with current understanding of SOX10 function in the affected cell types identified through in vivo and in vitro models. We also discuss research options to increase our understanding of the origin of the observed phenotypic variability and improve the diagnosis and medical care of affected patients.
- human genetics
- mutation
- nervous system diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SOX10 belongs to the SOX family of transcription factors, of which the members are defined based on the presence of a 79 amino acid DNA-binding domain with homology to the high mobility group (HMG) box of SRY (sex-determining region Y; hence SOX, Sry bOX). These factors are involved in multiple developmental processes, such as male differentiation, skeletogenesis, neurogenesis and neural crest (NC) development, where they control stemness, cell fate and differentiation.1–4 The growing number of developmental disorders associated with mutations in SOX genes underscores their importance during development.5 The SOX10 transcription factor is a characteristic marker for migratory multipotent NC progenitors as well as for various NC derivatives.
The NC is a specific population of cells in vertebrates that arise at the edge between the neural and non-neural ectoderm, delaminate from the dorsal aspect of the neural tube, and migrate through several routes to reach target tissues and give rise to neurons and glia of the peripheral nervous system (PNS), including sensory, autonomous and enteric ganglia, Schwann cells and olfactory ensheathing cells, melanocyte pigment cells, skeletal structures and mesenchyme of the head, face and neck, outflow tract of the heart, and smooth muscle cells of the great arteries.6 7
Over the years, heterozygous SOX10 mutations have been associated with various phenotypes that extend beyond Waardenburg syndrome (WS; depigmentation features and deafness) and Hirschsprung disease (HSCR; intestinal aganglionosis) initial diagnosis. Here, we present an up-to-date overview of these various clinical manifestations, along with our current understanding of how they are explained by SOX10 dysfunction in several NC derivatives and extra-NC tissues (inner ear and oligodendrocytes), and of the origin of phenotypic variability.
SOX10: structure and regulation of the gene, protein domains and post-transcriptional modifications
The human SOX10 and mouse Sox10 genes encode an open reading frame of 466 amino acids that share 92% nucleotidic and 98% amino acid sequence identities.8 The absence of a complete description of the human gene 5’ non-coding exon(s) has given rise to two coexisting exon numbering systems. Historically, exons 1 and 2 are non-coding, the initiation codon is found in exon 3, and the stop codon in exon 5.8 The second system is based on the reference transcript NM_006941, with only one non-coding exon in the 5’UTR (untranslated transcribed region) and a total of four exons. A major transcript of ~3 kb is detected in most tissues tested, consistent with the predicted SOX10 mRNA sequence.9 10
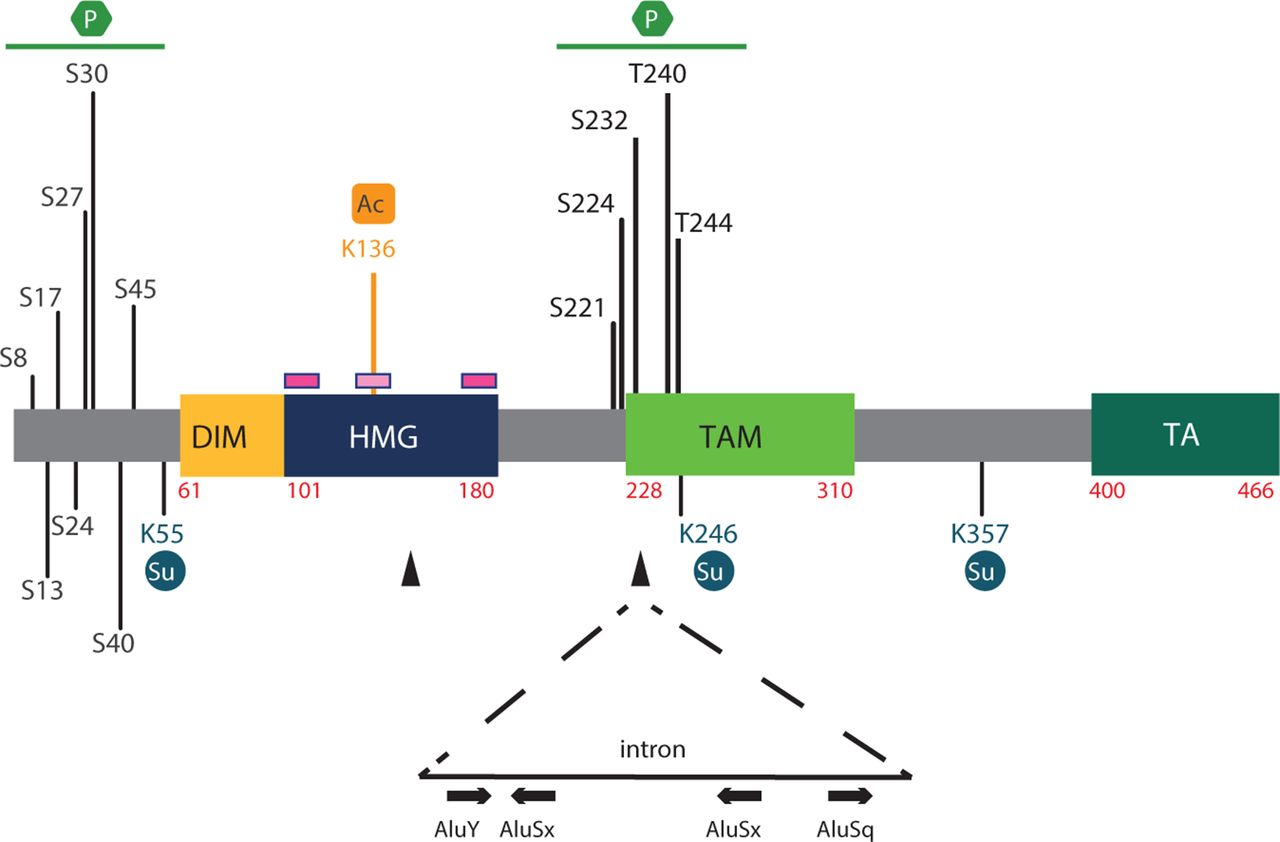
The protein’s structure is schematised in figure 1. As for all other members of the SOX family, the previously mentioned HMG domain forms an L-shaped module composed of three alpha helices that bind to DNA sequences in the minor groove (matching or resembling C[A/T]TTG[A/T][A/T]), bending the DNA molecule and interacting with other proteins to establish stable and active transcriptional complexes3 4 (the most recent model can be found in Haseeb and Lefebvre11). This domain also harbours two nuclear import (nuclear localisation signal) and one export (nuclear export signal) signals.12 13

Schematic of the SOX10 protein and post-translational modifications. Domains of human SOX10. The numbers refer to amino acid residues. The pink lines above the HMG domain represent the NLS sequences, one at each end of the HMG domain, and the light pink line the NES sequence. Note that although nucleocytoplasmic shuttling of the protein has been well documented for several SOX factors,12 13 in vivo regulation of SOX10 through nuclear translocation is yet to be clarified. Black arrowheads represent the localisation of junctions between exons. Post-transcriptional modifications, including acetylation (Ac), phosphorylation (P) and sumoylation (Su), are indicated, along with the position of modified amino acids. Note that a putative acetylation site was identified in SOX2 and is conserved in SOX10.17 Sumoylation by Ubc9 occurs at lysines K55, K246 and K357 with consequences on cell fate decision.19 20 Mechanistically, sumoylated SOXE proteins fail to interact with the coactivator CBP (CREB-binding protein)/p300 and instead recruit the GRG4 corepressor (Groucho-related protein 4/TLE4, transducing-like enhancer of split 4), leading to strong inhibition of SOXE target genes.106 Among the identified phosphorylation sites, note that ERK phosphorylates T240 and T244, inhibiting the sumoylation of SOX10 at K55 and transcriptional activity.107 Additional phosphorylation sites have been described from large-scale proteomic screens in melanoma, breast tumours and mouse neuroblastoma (serine S8, S13, S17, S24, S27, S30, S40, S45, S221, S224 and S23216). The relevance of most has not been functionally assessed. SOX10 phosphorylation sites are localised in two distinct clusters, one at the amino terminus, 5’ of the dimerisation domain, and the other at the centre of the protein. FBXW7-mediated ubiquitination of SOX10 has also been shown to control protein stability. The region involves aa 235–244 of the human protein. A search of the public REDIportal (http://srv00.recas.ba.infn.it/atlas/) revealed various A-to-I editing sites located in AluY, AluSx or AluSq sequences embedded in the last SOX10 intron. In each Alu sequence schematised from left to right, the number of A-to-I sites identified in >10 samples in various tissues (including the brain, gut, nerves (tibial), breast and salivary glands)/total number of A-to-I modifications reported is: AluY: 67/224; AluSx: 25/87; AluSx: 31/92; and AluSq: 20/73. DIM, dimerisation domain; ERK, extracellular signal-regulated kinase; HMG, high mobility group domain; NES, nuclear export signal; NLS, nuclear localisation signal; TA/TAC, transactivation domain in C-terminal; TAM, transactivation domain in the middle of the protein.
SOX10 shares additional domains with SOX8 and SOX9, all three forming the SOX_E group (SOX factors have been subdivided in several groups based on the amino acid identity within their HMG domain) (figure 1). Among them, the dimeric domain (DIM) confers preferential binding of SOX_E members to target sites containing two inverted SOX motifs separated by three to four nucleotides and promotes homodimerisation or heterodimerisation through DIM:HMG interactions.14
Within its C-terminus, SOX10 contains a potent transactivation domain called the TA or TAC (transactivation domain in C-terminal).4 Another weaker and context-dependent transactivation domain has been identified in the middle of SOX10, the so-called K2 domain or TAM (transactivation domain in the middle of the protein), and was recently shown to synergise with TA/TAC in all SOX_E members.11 15
Various post-transcriptional and post-translational modifications modulate the activity, stability and intracellular localisation of SOX1016 (figure 1). Several of these modifications are inferred from those occurring in other SOX factors, as for the lysine K136 acetylation site.16–18 Others, including phosphorylation sites, were mainly found from large-scale proteomic screens performed in cancer cells. SOX10 sumoylation by UBC9 (sumo-conjugating enzyme UBC9) is the best described one. Occurring at lysines K55, K246 and K357,19 it inhibits NC development and promotes development of non-sensory cranial placodes in vivo.20 Absence of A-to-I RNA modification mediated by the ADAR (adenosine deaminase RNA-specific) enzyme family was recently reported to alter melanocyte and Schwann cell development.21 Examination of the public REDIportal shows that SOX10 is under such regulation in humans (but not mice).
Finally, several regulatory regions likely involved in driving SOX10/Sox10 expression have been identified using various cell lines and zebrafish or mice models (ref 22 and references therein). Methylation of the Sox10 promoter by DNA methyltransferase 3 has also been shown to arrest NC generation in chicks.23
Involvement of SOX10 in WS: role in melanocytes and enteric nervous system
The identification of Sox10 as the gene mutated in the spontaneous Dom mutant mouse (Dominant megacolon; intestinal aganglionosis, white belly spot and white paws) first shed light on the essential function of this transcription factor in NC development. In this strain, a Sox10 frameshift mutation results in alteration of binding to some DNA target sequences in vitro, of transactivation capacity and synergistic action with several cofactors.9 24–27 This observation immediately led to test SOX10 involvement in Waardenburg-Hirschsprung disease.8 Also known as WS type 4 (WS4) or Waardenburg-Shah syndrome, Waardenburg-Hirschsprung encompasses symptoms of WS and HSCR (Mendelian inheritance in man, MIM) #613266).28–30
HSCR is the most common enteric neuropathy, occurring in 1 of 5000 neonates, and is characterised by the absence of enteric ganglia from a varying length of the distal gut, leading to intestinal obstruction in neonates or severe constipation in adults (MIM #142623).29 30
WS is a genetic disorder characterised by sensorineural hearing loss (SNHL) and pigmentation anomalies, including depigmented patches of skin and hair and vivid blue eyes or iris heterochromia (MIM #193500). Four types of WS are clinically defined, based on additional features due to defects in structures mostly arising from NC derivatives: WS1 is further characterised by dystopia canthorum, WS3 by musculoskeletal abnormalities of the limbs, WS4 by HSCR, whereas WS2 has no further significant features. In addition to SOX10, four main genes are involved in WS thus far: MITF (melanocyte inducing transcription factor) in WS2, PAX3 (transcription factor paired Box 3) in WS1 and WS3, EDN3 (endothelin 3) in WS4, and EDNRB (endothelin receptor type B) in WS4 and WS2.28 31 32 SOX10 has been shown to regulate and interact with several of these genes.28 33
SOX10 screening in WS4 cases led to the identification of the first heterozygous mutations in 1998.8 In 2007, SOX10 mutations were shown to be also responsible for approximately 15% of WS2 cases.34 By contrast, SOX10 involvement in isolated HSCR is very limited. For example, screening of 229 isolated HSCR cases led to the identification of only one frameshift mutation inherited from an asymptomatic mother (germline mosaicism has been proposed).35
Certain patients with WS4 or PCWH (see later) present with hypoganglionosis or chronic intestinal pseudo-obstruction (CIPO) instead of HSCR.28 36–39 Given the role of SOX10 in enteric nervous system (ENS) development, CIPO is probably neurogenic. Aganglionosis is therefore not the only mechanism underlying the intestinal dysfunction in patients with SOX10 mutations.
Each of the clinical manifestations described above can be explained by dysregulation of SOX10 during melanocyte and ENS development. WS accounts for a developmental defect in both skin melanocytes and a melanocyte-derived cell lineage of the inner ear, called intermediate cells of the stria vascularis, necessary to the inner ear homeostasis.40 In melanocytes, SOX10 controls proliferation, survival and differentiation by directly and sequentially activating a number of downstream target genes.4 41–43 From the NC, a SOX10–PAX3 pair activates the expression of Mitf/MITF, which then acts as a SOX10 partner to activate the expression of Dct (dopachrome tautomerase) and Tyr (tyrosinase), both involved in melanocyte differentiation and melanin synthesis.27 32 42 44 45 In 2015, an extensive genome-wide catalogue of SOX10 targets was obtained.46 For the first time, integrated chromatin occupancy and transcriptome analysis suggested a role of SOX10 in both transcriptional activation and repression. SOX10 was also shown to cooperate with MITF to facilitate BRG1 (Brahma-related gene 1/SMARCA4, SWI/SNF related, matrix associated, actin-dependent regulator of chromatin) binding to distal enhancers of melanocyte-specific genes, promoting differentiation.47
In the developing gut, SOX10 is expressed in all NC-derived ENS progenitors.22 24 48–50 Later, SOX10 is maintained in enteric glia but downregulated in cells that are committed to neurons (see refs 25 50 for examples). Most publications suggest a role of SOX10 in the maintenance of enteric progenitors,22 49 and overexpression of SOX10 inhibits enteric neuron differentiation, without altering commitment to the neurogenic lineage.25 51 These cellular functions rely on the capacity of SOX10 to regulate (along with several cofactors) various target genes, including Ret (RET proto-oncogene; a receptor tyrosine kinase involved in ENS development and the main HSCR-related gene), Ednrb and Sox10 itself.22 33 52 53 As an example SOX10 and ZEB2 (zinc-finger E-box binding homeobox 2; a negative regulator of NC differentiation) both bind to Ednrb promoter-specific regions, highlighting the role of this ‘triade’ in controlling the maintenance of multi-potential enteric progenitors and their differentiation process.33
Hearing loss associated with SOX10 mutations: beyond melanocytes, SOX10 expression in inner ear and related deficits
SNHL due to SOX10 mutations, as for the other WS genes, is typically prelingual, non-evolutive, profound and bilateral. However, it can also be moderate and asymmetric or unilateral.
Aside from the intermediate-cell alterations mentioned above, inner ear malformations have been noted in some patients with WS long ago.54 It now appears that only patients with a SOX10 mutation present with these abnormalities: hypoplasia/dysplasia or agenesis of the semicircular canals and enlarged vestibules are very frequent, while agenesis of the vestibulo-cochlear nerve and cochlear deformities have also been reported.55–57 Consequently, temporal CT scan or MRI is of particular interest in diagnosis. In our experience, this feature is highly penetrant when interpreted by a specialised radiologist. However, recent papers reported the absence of imaging abnormalities in the inner ear of a few patients with SOX10 mutations. A complete exploration of the vestibular function has yet to be performed.
These observations are consistent with an expression profile of Sox10 in the ear. Sox10 is expressed in the placode-derived otic vesicle from E9.5 onward and then in the developing epithelium of the cochlea and vestibule, before being restricted to supporting cells of the neurosensory epithelium. Sox10/SOX10 promotes the survival of cochlear progenitors during formation of the otocyst and the organ of Corti, plays a role in glial development of the cochleovestibular ganglia, and its NC-targeted deletion leads to improper neuronal migration and projection.58–60 The resulting inner ear malformations differ depending on the animal model.58 61 62 RNA-seq studies of inner ear development in a pig model showed dysregulation of WNT1 (Wnt family member 1; a regulator of cell fate and patterning), KCNQ4 (potassium voltage-gated channel subfamily Q), STRC (stereocilin; a protein associated with the hair bundle of the ear sensory cells) and PAX6 (transcription factor Paired Box 6) networks.62
In agreement with this broad function, SNHL appears to be the most penetrant sign in cases of SOX10 mutation, leading to the observation that certain patients can present with isolated SNHL until minor signs are revealed on medical reinterview.63
PCWH and PCW phenotypes: important function of SOX10 in Schwann cells and oligodendrocytes
Beyond WS2 or WS4, neurological alterations have been identified in the so-called PCWH syndrome (peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, Hirschsprung disease; MIM #609136).28 36 64 Depending on the severity of myelin defects in the PNS and central nervous system (CNS), patients with PCWH exhibit variable symptoms that often include delayed motor and cognitive development, cerebral palsy, ataxia, spasticity, congenital nystagmus, hyporeflexia, distal sensory impairments and distal muscle wasting. This phenotype is recapitulated in a transgenic mouse model with several copies of SOX10 carrying the first PCWH mutation described65 66 and is mostly explained by the role of SOX10 during differentiation of myelinating Schwann cells and oligodendrocytes, both ensuring rapid salutatory conduction along axons.67 68
In the PNS, SOX10 controls each differentiation step by inducing stage-restricted transcriptional regulators, which are then recruited as partners to activate specific sets of target genes, allowing progression to the next stage.67–72 For example, in immature Schwann cells, SOX10 induces the expression of OCT6 (POU3F1, POU class 3 homeobox 1). Both factors then cooperatively activate the programme required for progression into the promyelinating stage. Their target EGR2 (early growth response 2) then associates with SOX10 to activate the myelination programme.
In the CNS, analyses of various animal models revealed an essential role of SOX10 in the terminal differentiation of oligodendrocytes in coordination with OLIG1 (OLIGodendrocyte transcription factor 1), MYRF (myelin regulatory factor), TCF4 (transcription factor 4, which has an important role in CNS development) and CHD7 (chromodomain helicase DNA-binding protein 7; the gene involved in CHARGE syndrome (Coloboma, Heart anomaly, choanal Atresia, Retardation, Genital and Ear anomalies)).68 Many genes that are activated during terminal differentiation of oligodendrocytes are direct targets of SOX10, but there are only few known SOX10 targets in oligodendrocyte precursors.68 73 74 Recently, MYRF was identified as a decisive factor that helps SOX10 to switch between its target genes along oligodendrocyte differentiation process.75
Of interest, some of the genes directly regulated by SOX10 in PNS and CNS are known to be responsible for hypomyelinating/demyelinating diseases, with some described mutations in these genes that directly result from a loss of regulation by SOX10.76–78
Involvement of SOX10 in Kallmann syndrome and its role in olfactory ensheathing cells
SOX10 was considered to be a candidate gene for Kallmann syndrome (KS, hypogonadotropic hypogonadism and anosmia; MIM #308700) based on the unexpected high frequency of olfactory bulb agenesis55 associated with rare clinical reports of hypogonadism or anosmia in patients with WS/PCWH with a SOX10 mutation. The screening of cohorts indeed revealed SOX10 mutations in patients with KS, most of whom also have hearing impairment.79 Since then, many other SOX10 mutations have been characterised in KS or normosmic idiopathic hypogonadotropic hypogonadism (nIHH), although they were usually not functionally characterised and a subset of them appeared unlikely to be pathogenic (see Review of SOX10 variations). Interestingly, KS and WS are not mutually exclusive, and some patients with an initial diagnosis of WS have been further diagnosed with hypogonadism at puberty.80 We believe that anosmia and hypogonadism are still underestimated in patients with WS with a SOX10 mutation, as signs of KS are difficult to diagnose before puberty. Of note, in the absence of pigmentary disturbances, the association of KS+hearing impairment+abnormalities of the semicircular canals can lead to a differential diagnosis with mild forms of CHARGE syndrome (MIM #214800) (examples in online supplemental table 1).
Supplemental material
The common cause of anosmia and hypogonadism is a defect in a developmental sequence of GnRH (gonadotropin-releasing hormone) neurons migrating along the peripheral olfactory nerve up to and through the olfactory bulb. In the Sox10 knockout mouse, a primary defect of the olfactory ensheathing cells leads to a secondary defect of the olfactory nerve pathway, defasciculation and misrouting of the nerve fibres, impaired migration of GnRH cells along this route, and disorganisation of the olfactory nerve layer of the olfactory bulbs.79 Dysregulation of the frizzled related protein FRZB may contribute to explain the defect in olfactory axon targeting but not GnRH neuron migration.81
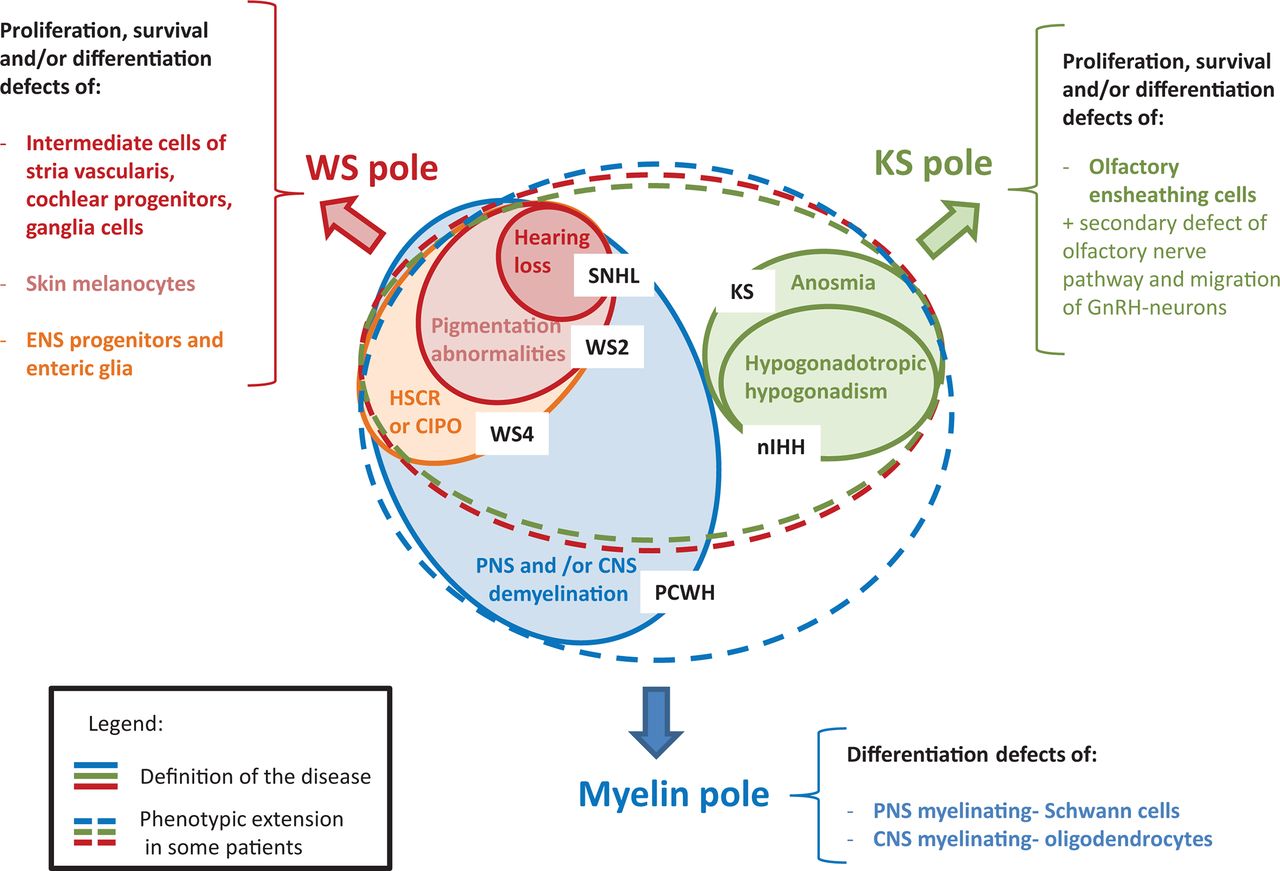
A summary of the recurrent clinical manifestations due to constitutive SOX10 mutations along with affected cell types is presented in figure 2.

Summary of the clinical spectrum due to SOX10 mutations and the corresponding SOX10 function(s). The picture is organised around three clinical poles that correspond to different diagnosis entries. The WS pole is indicated in red, the myelin pole in blue and the KS pole in green. The plain line corresponds to the definition of the disease, while the dotted lines indicate the main clinical extension of these syndromes in case of SOX10 mutation. Note that the area of the circles is not proportionate to the relative frequency of each syndrome (for an idea about the number of patients, see figure 3B). CIPO, chronic intestinal pseudo-obstruction; CNS, central nervous system; ENS, enteric nervous system; GnRH, gonadotropin-releasing hormone; HSCR, Hirschsprung disease; KS, Kallmann syndrome; nIHH, normosmic idiopathic hypogonadotropic hypogonadism; PCWH, peripheral demyelinating neuropathy, central dysmyelination, Waardenburg syndrome, with Hirschsprung disease; PNS, peripheral nervous system; SNHL, sensorineural hearing loss; WS, Waardenburg syndrome; WS2, Waardenburg syndrome type 2; WS4, Waardenburg syndrome type 4.
Involvement in cancer, sex reversal, associations and reports of the first biallelic mutations
Beyond congenital disorders, a role of SOX10 in cancer progression has been reported. SOX10 protein is highly expressed in breast, glioma, glioblastoma, salivary adenoid cystic tumours and hepatocellular carcinoma (see The Cancer Genome Atlas). The association of SOX10 with melanoma is the best described, but only a limited number of variants have been reported so far.82–84
Several reports of duplications in the 22q13.1 region have been published that may include one or several signs of WS/PCWH and sex reversal in a number of cases.85 Sex reversal has been suggested to be due to the overexpression of SOX10, consistent with observations in a Sox10 transgenic mouse model.86 However, we found a SOX10 triplication (four doses of SOX10 instead of two) in a 47,XX baby girl without sex reversal (online supplemental table 1), indicating that overexpression of SOX10 alone may not be sufficient, the sign is not fully penetrant or the overexpression of other genes has the opposite effect, depending on the size of the rearrangement.
More complex and questionable associations have also been described. For example, increased DNA methylation of SOX10 has been linked to oligodendrocyte dysfunction in patients with schizophrenia.87
Two cases of biallelic SOX10 deletion have been characterised and, although not reported in the papers, they appear to represent the first and second pregnancy from the same couple.88 89 Both parents are heterozygous for one of the two SOX10 deletions and present with a classic form of WS. Biallelic SOX10 loss-of-function results in a severe polymalformative fetal phenotype. Eighteen other genes were included in the maternal deletion and may participate in the phenotype.
Finally, the development of large gene panels for diagnosis and whole exome/whole genome sequencing has led to SOX10 mutations being found in unexpected contexts. A number of cases have thus been listed in cohorts of neurodevelopmental defects, hearing impairment and endocrinological problems. Due to the diverse phenotypes related to SOX10 mutations, making sense of such findings can be challenging.
Review of SOX10 variations
During the first 15 years after their discovery, most SOX10 disease-associated point mutations were shown to result in premature termination codons, with strikingly few exceptions.28 Missense mutations started to be found simultaneously with the finding that SOX10 mutations can cause less severe syndromes than life-threatening WS4 or PCWH.90
An up-to-date summary of confident mutations of SOX10 (approximately 300 independent cases, including unpublished ones in online supplemental table 1) is presented in figure 3A. Truncations (stops, frameshifts) are found in slightly more than half of all cases. Approximately one-third of all mutations are non-truncating variations, either missense or small inframe insertions/deletions, the rest being either complete or partial copy number variations of the gene (approximately 10%) and rare mutational mechanisms (splice mutations, mutation of the initiation codon or non-stop mutations (five cases to date)). Truncating mutations can be located anywhere, except in the very extreme C-terminus. On the contrary, missense mutations are tightly clustered in the DNA-binding domain (HMG), a frequent finding for transcription factors. We have thus far found no specific link between SOX10 missense mutations and residues involved in post-translational modifications.

{kind=link}
{kind=link}
{kind=link}
Review of SOX10 mutations. (A) Representation of SOX10 truncating and non-truncation mutations along the SOX10 protein. We made a list of all published SOX10 mutations, starting with the LOVD database that we curated up to 2015 (https://databases.lovd.nl/shared/genes/SOX10), updated with the literature and finally completed using the HGMD (Human Gene Mutation Database) professional database (https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database) for a few mutations that were reported in cohorts of unspecific diagnosis and would have been missed by common keywords. We prioritised the strength of the data to create this figure, as our goal was not to include all cases but to provide a reliable picture of the SOX10 mutational spectrum: we retained papers for which the data allowed curation and removed neutral variants and variants of unknown significance, duplicated patients or publications, and publications with inconsistent findings. Finally, we added our unpublished cases (listed in online supplemental table 1). ‘M1?’ indicates a mutation of the initiation codon (p.Met1?). (B) Proportion (in percentage) of the different types of mutations for each syndrome. ‘n’ indicates the number of independent cases included in each group. (C) Localisation of the truncating (stop and frameshift) mutations along the SOX10 protein associated with each phenotype. Note that (1) the phenotypic description was sometimes too incomplete for inclusion in B and C; (2) among the familial cases showing intrafamilial differences in phenotype, we considered the phenotype of the index case; (3) KS/nIHH is given regardless of the presence of WS signs or not, and anosmia without hypogonadism was not considered; and (4) because presence or absence of a demyelination is frequently unreported or not evaluated, we conserved all the patients with neurological features in the PCW/PCWH group when the data seemed consistent. DIM, dimeric domain; HMG, high mobility group; KS, Kallmann syndrome; LOVD, Leiden OPen Variation Database; nIHH, normosmic idiopathic hypogonadotropic hypogonadism; PCW/PCWH, peripheral demyelinating neuropathy, central demyelination, Waardenburg syndrome, with or without Hirschsprung disease; TA, transactivation domain in C-terminal; TAM, transactivation domain in the middle of the protein; WS, Waardenburg syndrome; WS2, Waardenburg syndrome type 2; WS4, Waardenburg syndrome type 4.
Of course, rarity in control populations is not sufficient to confer pathogenicity and the prediction of pathogenicity by dedicated tools is of indicative value only. Among the published SOX10 missense variations that are located outside of the HMG domain, most should be considered variants of unknown significance. From our experience and bibliography review, it appears that extremely rare or new missense variations have a high probability of being truly pathogenic if located in the HMG domain, whereas missense mutations located outside of this domain, even if rare and ‘predicted pathogenic’ by in silico tools, are less likely to be pathogenic and should be considered cautiously. We have worked on SOX10 since its characterisation, both in the research and clinical context, and have only once found an exception to each of these rules. With the increasing number of mutations described, it appears that there may be a second, rare spot of mutations in the dimerisation domain (three variations reported in four independent cases, creating or removing valines at residues 76, 79 and 80), although functional tests are required to reach a definitive conclusion91 92 (online supplemental table 1).
Due to the well-documented incomplete penetrance and digenism in KS and nIHH, there is a tendency in the literature to overevaluate the pathogenic probability of rare variants. Certain SOX10 variations have been considered to be pathogenic or likely pathogenic without many arguments (low pathogenicity scores, no functional tests, proven not pathogenic in another paper, inherited from healthy parents or without segregation study, and/or associated with an obvious causative mutation in another KS/nIHH gene). On careful review, we consider several of these rare missense variants to more likely be neutral (although some still may be hypomorphic variants exerting their effect on a multigenic background, but this has thus far not been proven) and did not include them in figure 3.
In early studies, most SOX10 variants were found to be de novo, which was thought to be due to the severity of HSCR in WS4. Given the cumulative number of WS2 cases now described, the life-threatening hypothesis cannot completely explain the proportion of de novo mutations. The possibility of hypogonadism in patients probably also contributes to this observation. However, the proportion of familial cases has tended to increase over the years and now represents approximately 20% of cases. These cases revealed an important intrafamilial phenotypic variability. Several mutations have now been found in independent cases and also show interfamilial phenotypic variability. Parental mosaicism is found in approximately 3%–4% of cases, but a recent study reported a higher proportion in a small series using more sensitive methods.93
The proportion of mutations relating to phenotype is summarised in figure 3B. There is a large proportion of truncating mutations in WS4 and PCWH. The proportion of missense increases in WS2 and even more strongly in KS. Thus, not all missense mutations may be null mutations.
The location of truncating mutations along the gene (figure 3C) confirms the correlation between PCWH and the escape from non-sense-mediated RNA decay (NMD) (mutations located in the last coding exon and last 45 nt of the penultimate exon).64 Of note, some of the cases that appear not to respect the NMD rule may be misclassified (whether demyelination is proven or not is not always reported). The severity of PCWH was shown to be linked to the location of the mutation within the last exon: the earlier the truncation, the more severe the phenotype.64 This tendency is visible in the graphs (compare the truncating mutations of WS vs PCWH in the last exon). A few cases escape the rule, with no clear explanation so far.
Finally, heterozygous deletions/duplications can be intragenic or lead to complete gene loss and be as large as several Mb, encompassing other genes and leading to more complex phenotypes.
Functional consequences of SOX10 mutations and the origin of phenotypic variability
Most in vitro functional tests found in the literature rely on the ability of SOX10 to activate its target genes, alone or in combination with its cofactors. The construct most frequently used is a luciferase reporter under the control of the MITF-M (melanocyte-specific isoform) promoter. Additional targets, immunohistochemistry and assessment of the contribution of the DNA-binding capabilities have sometimes enriched such studies.
Functional analysis of the first SOX10 missense mutation suggested that differential tissue-specific gene regulation could account for the phenotype observed in patients.94–96 Since, many SOX10 missense mutations associated with a variety of phenotypes, ranging from WS2 to WS4 and PCWH, have been tested, but no clear correlation between in vitro results and the phenotypes could be established.79 90 97
The development of in vivo tests is therefore required to facilitate the establishment of genotype–phenotype correlations. The only model currently published is in ovo chick electroporation in the developing neural tube.26 However, the effect of most of the mutations on early NC development precludes the analysis of their role in later developmental processes. Use of an inducible model would be of interest. Alternatively, zebrafish or the use of induced pluripotent stem cells differentiated towards NC derivatives of interest could be future models of choice.
As mentioned earlier, the presence or absence of a neurological phenotype that characterises PCWH or PCW was proposed to be related to NMD.64 The proposed mechanism is that mutant proteins that have escaped degradation via the NMD pathway result in dominant-negative activity that impairs the function of the wildtype SOX10 allele and lead to PCWH, while those located in the first coding exons activate the NMD RNA surveillance pathway, leading to degradation, haploinsufficiency and a classic WS phenotype.
However, several non-stop mutations have also been described to be associated with a PCW/PCWH phenotype. This is thought to be due to the generation of a specific inframe new C-terminus generated by the loss of normal termination. Functional studies of an equivalent mouse mutant allele showed that the additional 82 amino acids contain a deleterious (tryptophan-arginine (WR) domain, supporting a toxic gain-of-function.98 This is consistent with the recent report of a frameshift mutation that also elongates the protein, but in a different reading frame does not lead to PCWH (p.Tyr460Leufs*42).99 The observation of another, transgenic mouse model carrying different copy number variations of the first described SOX10 non-stop mutation suggested PCWH is due to a dominant-additive, rather than dominant-negative, effect.66 Finally, duplication of the 22q13.1 region, including SOX10, can also induce PCWH,85 supporting the hypothesis that it is promoted by a gain-of-function rather than a dominant-negative effect. Regardless of the mechanism, these observations all indicate that NC derivatives are highly sensitive to the dose of SOX10 and its function.
SOX10 expression is regulated by numerous enhancers. It is thus possible that certain cases with minor expression could also be due to specific dysregulation of one or a subset of such enhancers. This paradigm is supported by disruption of tissue-specific, long-distance regulatory regions of SOX9 causing endophenotypes.100 101 A large de novo deletion encompassing three SOX10 regulatory elements has been characterised in a patient with typical WS4,102 leading to the hypothesis that variations affecting certain identified regulatory sequences could be the cause of unexplained WS2 or isolated HSCR. Screening for mutations in SOX10 regulatory regions in WS2 turned out to be unfruitful.103 On the contrary, one deletion and two point variations affecting binding sites for known NC transcription factors were identified in 3 of 144 cases of isolated HSCR, both variations being in association with a HSCR-predisposition polymorphism at the RET locus.104 With the implementation of population databases, it now appears that one of these two variants is less rare than expected (22-38016774 G-C; about 1/1000 according to the gnomAD database; https://gnomad.broadinstitute.org/), which questions its involvement. These results are yet to be replicated for a pertinent interpretation.
In any case, in vitro/in vivo tests will not be able to explain all phenotypes, as phenotypic variability is commonly recognised in patients with SOX10 mutations, even those with the same mutation and even within the same family. This suggests that the genetic background is influential, as has often been suggested for HSCR.53 105 Because the identification of modifier genes has been hampered by the small number of available patients, most modifier gene studies have relied on Sox10 mouse models.22
Despite such variability, certain specificities have been reported for a few peculiar missense mutations. Here, we want to discuss the case of the Gln174/Pro175 missense mutations. The observation that certain SOX10 missense mutants accumulate in nuclear foci in transfected cells, where they colocalise with p54NRB (nucleo ribo binding protein, 54 kDa/NONO, non-Pou domain containing octamer binding; a multifunctional protein known to be a marker of ‘paraspeckles’), leads to characterise missense mutations of amino acids 174 and 175 as associated with a peculiar phenotype (refs 79 97 and unpublished data (S. Marlin, N. Bondurand and V. Pingault, 2016)). Remarkably, the cotransfection of foci-forming mutant with wildtype constructs led to the sequestration of wildtype SOX10 in these ‘foci’ and altered the synergistic activity of SOX10 and p54NRB. A dominant-negative effect was therefore proposed to contribute to or be at the origin of the progressive central and peripheral neurological phenotypes observed in patients carrying these specific missense mutations and may thus be the basis of a hitherto unexplored molecular mechanism for genotype–phenotype correlations. These data need to be confirmed in more physiological models.
The phenotype variability finally leads to question the risk of a more severe phenotype in cases of recurrence in a family. The risk of the PCWH phenotype after a first non-PCWH case is considered to be low. On the contrary, there is a risk of WS4 after a first, milder case of WS2. This situation has been reported several times; however, a bias in the representation of these cases in the literature can be expected, as a second mildly affected member is less likely to result in a visit of the family to the geneticist’s office, molecular analysis and ultimately publication. As a result, the true risk is difficult to quantify.
Conclusion, with a few tips to help in variant classification
During twenty years of cases and cohorts reporting, SOX10 variants have been involved in WS2/WS4/PCWH/Kallmann syndrome/pseudo-isolated hearing loss/HSCR or CIPO and any combination. This is correlated with the known developmental functions of SOX10. All these phenotypes should be considered as a clinical continuum with variable expression, rather than as independent diseases, conferring a mild to life-threatening syndrome. Observation of the familial cases and of a few recurrent variations documented a high phenotypic variability, even within a single family.
Most mutations predict a truncation of the protein, but the proportion of missense variations has increased with time. Missense variations (or small in-frame insertions/deletions) outside of the HMG domain should be considered with caution, even with good in silico pathogenicity scores. The fact that the variation is already published can be used as a supporting argument only if the strength of the published data has been verified (also a general recommendation of the American College of Medical Genetic).
The most (almost fully) penetrant sign observed in patients is hearing impairment. Pigmentation defects are not always present. Confirmed incomplete penetrance appears to be very rare, but a targeted clinical reevaluation may be necessary to assess mild signs. Searching for inner ear-specific malformations by imaging is highly informative. The absence of olfactory bulbs could be investigated at the same time by MRI. The only strong phenotype-genotype correlation usable in phenotype prediction, thus far, is the link between NMD escape and PCW/PCWH.
Ethics statements
Patient consent for publication
Ethics approval
Ethics approval is not applicable. This study does not involve human participants in a research study. Only mutations found on a diagnosis basis are reviewed in a retrospective manner (list of mutations along with scarce clinical information).
Acknowledgments
We apologise to all whose contributions were not cited due to space limitations.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was published online. The conclusion has been added, and figure 2 and the supplementary files have been replaced.
Contributors VP and NB: conceptualisation, literature review, data extraction, supervision and writing of the manuscript. LZ and WB-T: literature review, data extraction and editing of the manuscript. All authors have reviewed the manuscript and have approved its final version for publication.
Funding Our work is supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), Assistance Publique des Hôpitaux de Paris (AP-HP), funding from the Agence Nationale de la Recherche under the 'Investissements d’avenir' programme (ANR-10-IAHU-01), the MSD-Avenir fund (DEVO–DECODE project), Fondation Maladies Rares, and the European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreement (NeuCrest no: 860635).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.