Article Text

Abstract
Background Congenital nemaline myopathies are rare pathologies characterised by muscle weakness and rod-shaped inclusions in the muscle fibres.
Methods Using next-generation sequencing, we identified three patients with pathogenic variants in the Troponin T type 1 (TNNT1) gene, coding for the troponin T (TNT) skeletal muscle isoform.
Results The clinical phenotype was similar in all patients, associating hypotonia, orthopaedic deformities and progressive chronic respiratory failure, leading to early death. The anatomopathological phenotype was characterised by a disproportion in the muscle fibre size, endomysial fibrosis and nemaline rods. Molecular analyses of TNNT1 revealed a homozygous deletion of exons 8 and 9 in patient 1; a heterozygous nonsense mutation in exon 9 and retention of part of intron 4 in muscle transcripts in patient 2; and a homozygous, very early nonsense mutation in patient 3.
Western blot analyses confirmed the absence of the TNT protein resulting from these mutations.
Discussion The clinical and anatomopathological presentations of our patients reinforce the homogeneous character of the phenotype associated with recessive TNNT1 mutations. Previous studies revealed an impact of recessive variants on the tropomyosin-binding affinity of TNT. We report in our patients a complete loss of TNT protein due to open reading frame disruption or to post-translational degradation of TNT.
- diagnosis
- neuromuscular diseases
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Congenital nemaline myopathies (NMs) are rare muscle diseases characterised by abnormal muscle tone and the presence of rod-shaped inclusions in the muscle fibres. These rods are sometimes visible with light microscopy and Gomori trichrome staining. The rods are located mainly in the sarcoplasm and more rarely inside the nucleus.1 They are more easily visualised and authenticated by electron microscopy: they are composed of lattice-like filaments, resembling Z-streaks, and are often observed within disorganised myofibrillar networks.2 3 The location of the rods in the fibre, their ultrastructural appearance and the fibre type that preferentially contains them are all criteria that provide a genotype orientation. For example, the presence of intranuclear rods indicates that the alpha-actin-1 (ACTA1) gene has been affected, whereas a preferential location in the centre of the sarcoplasm more strongly suggests involvement of the Troponin T type 1 (TNNT1) gene.1 4 At least 13 genes are currently known to be involved in NM. They encode the thin filament proteins of the skeletal muscle sarcomere (nebulin, actin alpha 1, beta-tropomyosin-2, alpha-tropomyosin-3, troponin T type 1, cofilin-2, leiomodin-3 (LMOD3), skeletal troponin-3 and myopalladin), the Kelch domain-associated proteins (KBTBD13, KLHL40 and KLHL41) and a myosin whose function is unknown (MYO18B). The ryanodine receptor-3 gene has also been described in NM.5 Genotyping is now easier with next-generation sequencing (NGS)6 techniques as variants can be detected, including small deletions and insertions and variations in the number of copies.
The involvement of the TNNT1 gene has been exceptionally reported in various populations. Troponin T is one of the three subunits of the troponin complex7 and has been called the ‘slow skeletal muscle isoform of troponin T (TNT)’ because of its presence in slow skeletal muscle fibres. This protein is responsible for the link between the troponin complex and tropomyosin (Tm), which helps to regulate muscle contraction.8 Patients with NMs related to the mutations in the TNNT1 gene show phenotypical variability. Initially, this gene was implicated in the so-called Amish NM, which affects consanguineous Amish families,4 and linked to a nonsense mutation in exon 11 of TNNT1. Since then, 13 other patients with NM have been described, implicating the TNNT1 gene with autosomal recessive inheritance: 3 Dutch patients9 from the same family, 1 Hispanic patient10 and 9 Palestinian patients2 from seven different families. More recently, a new heterozygous missense mutation of TNNT1 c.311A>T; p.E104V was described,11 with NM inherited in an autosomal dominant manner.
We describe three new patients with NM due to recessive TNNT1 mutations identified by NGS and try to better specify the phenotypical spectrum and to identify the consequences of mutations on TNNT1 mRNA and protein levels.
Materials and methods
Patients were recruited through the French national network of neuromuscular diseases reference centres. Patients or parents gave informed consent for the genetic analysis according to French legislation.
Sequencing
Sanger sequencing of TNNT1 coding exons was performed for patient 3.
NGS was performed on extracted DNAs using customised designs of myopathy genes, as previously reported.6
An in-house bioinformatics spreadsheet was implemented for the copy number variation analyses, as previously reported.6 This was used to analyse the intersample normalised depth of coverage per exon in a given run. cDNAs of the TNNT1 transcripts were analysed on mRNA extracted from muscle biopsies; the total transcripts were amplified and sequenced after fragmentation and library preparation (NEBNEXT New England Biolabs). The library was then sequenced on a PGM IonTorrent platform. Transcript sequences and splice junctions were analysed using RNASTAR software.12
Western blot
The amount of skeletal muscle-specific troponin T (sTNT) in the muscle sample biopsies was determined by western blot analysis using antibodies directed against sTNT (Mab- CT3) with tubulin (Mab- Tub V.2.1) as loading protein sample control. Muscle homogenates were prepared from frozen muscle biopsy using Minilys homogeniser as described previously.13 Twenty micrograms of muscle homogenates were separated on a 12% acrylamide gel (Biorad) and transferred on immobilon as described previously.13 The patients and an age-matched (3.5 years old) non-affected control sample were tested.
Results
Patient 1
This 27-month-old patient was born full-term after a normal pregnancy to non-consanguineous parents from the Basque region of France. She was eutrophic and showed no difficulty in adapting to extrauterine life. No specific clinical signs were noted at birth. She had one older brother. Axial and peripheral hypotonia was observed at 2 months, associated with facial hypomimia, ‘fishmouth’, non-specific painful manifestations and tremor. Respiratory insufficiency appeared gradually, with secondary thoracic dystrophy, requiring ventilatory support by nocturnal non-invasive ventilation at the age of 16 months. She had areflexia of the four limbs. At 17 months, she showed axial weakness and inability to hold her head up or maintain a sitting position, contrasting with better distal mobility. Asymmetrical cervical stiffness was observed early on, as well as a limitation in hip abduction and knee retraction. A rapidly progressing kyphoscoliosis required treatment with a rigid brace from 20 months onward. Failure to thrive prompted the placement of a gastrostomy tube for enteral nutrition at 20 months. Her cognitive abilities were normal. Death occurred suddenly at 29 months. Creatine phosphokinase (CPK) testing, echocardiography, electroneuromyography and an MRI cerebrospinal flow study showed no abnormalities. There was no phrenic palsy. The muscle biopsy performed at 15 months revealed a clear disproportion in fibre size, with type I fibres consistently smaller than type II fibres, the dissolution of the myofibrillar network in the centre of certain fibres, early endomysial fibrosis, and rare vacuolated degenerative fibres. Ultrastructural analysis revealed a large number of rods located in the centre of the fibres in areas of sarcomeric disorganisation (figure 1). NGS of the exons and exon–intron junctions of the genes involved in congenital myopathies and muscular dystrophies revealed a homozygous deletion of exons 8–9 of the TNNT1 gene (figure 2). The boundaries of the deletion were determined by genomic sequencing (hg19chr2: g.55650633_55652981del and NM_003283.5: c.192+244_388-1191del).1 This deletion, predicted to be in frame, removed part of the functionally important Tm-binding site 1 domain (figure 3). The family segregation analysis concluded that the child was homozygous for this variation since she had inherited a mutated allele from each of her parents (table 1).
In-house bioinformatics script results for TNNT1 CNV familial segregation analysis
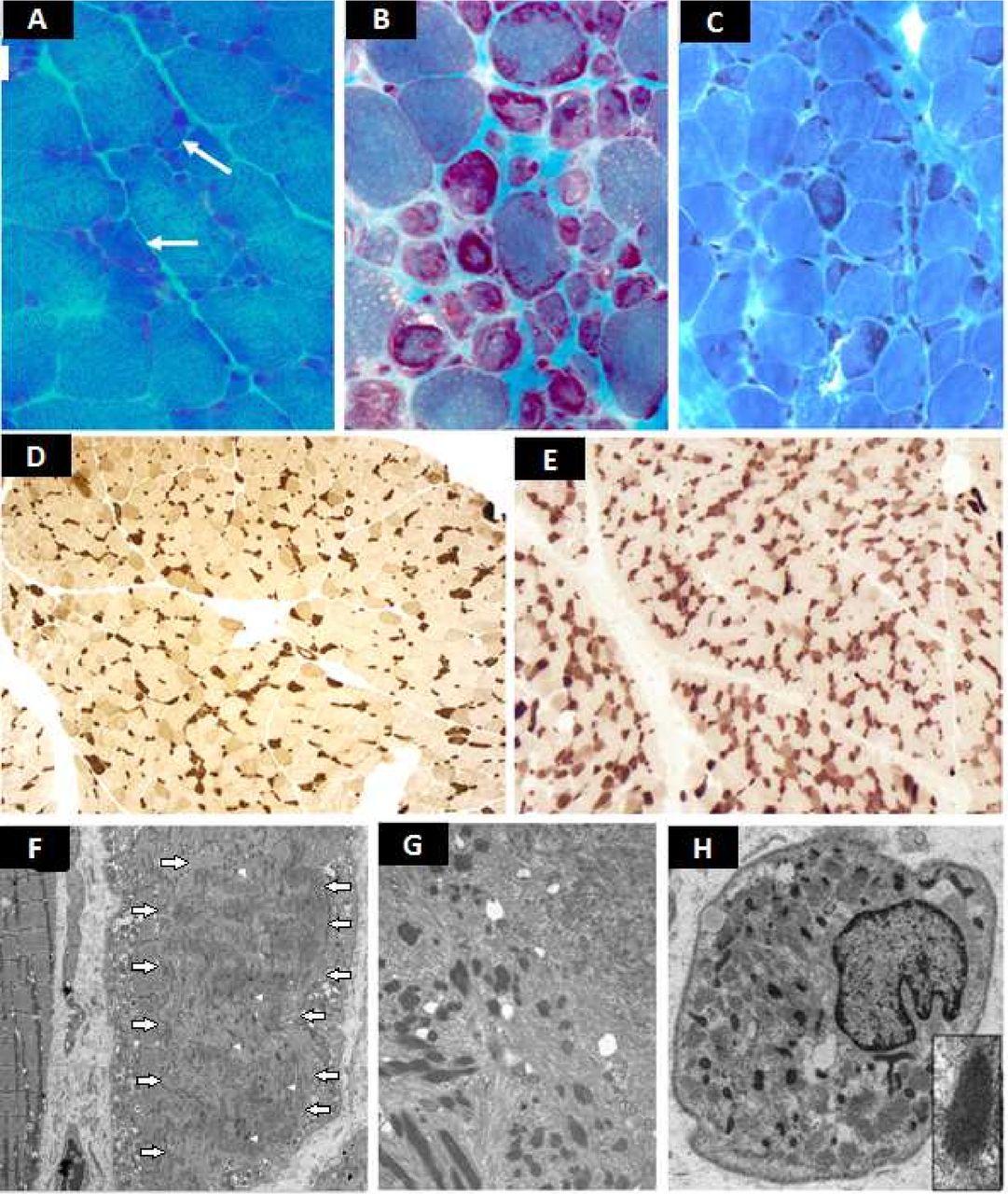
Representative frozen sections (A–E) and electronic microscopy images (F–H). Gomori trichrome: patient 1 (A): marked fibre size irregularity. Rare atrophic fibres with hyaline core-like intracytoplasmic inclusions (white arrows), slight increased endomysial connective tissue. Patient 2 (B): numerous rods and endomysial fibrosis. Patient 3 (C): rods are visualised in disorganised fibres. ATPase pH 4.6: patient 1 (D) and patient 3 (E): type I fibres are dark; type IIA fibres are pale, type IIB are intermediate stained; type I fibres are irregular and smaller than type II. No myopathic grouping. The proportion of type I and type II is preserved. No fibre-type grouping. Ultrastructure: patient 1 (F): the sarcomere structure is disrupted in a large circumscribed central area of a muscle fibre (fibre on the right, white arrows showing the outlines of this area). Rods are visualised within this area (white arrow heads showed some rods). Detail: a typical rod with homogeneous lattice Z disk-like structure in continuity with thin filaments. Patient 2 (G): numerous electron dense rods in a disorganised area. Patient 3 (H): rods are observed throughout the cytoplasm. Detail of a typical rod with lattice structure.

Homozygous deletion of exons 8–9 of the TNNT1 gene. Integrative Genomics Viewer visualisation of the deletion identified by NGS in case1 (I401). Precise breakpoint determination was further obtained by Sanger sequencing (hg19chr2 : g.55650633_55652981del ; NM_003283.5 : c.192+244_388-1191del). NGS, next-generation sequencing; TNNT1, troponin T type 1.

TNNT1 variants identified in patients. Patient 1 revealed a homozygous deletion of exons 8 and 9 of the TNNT1 gene. These two exons encode part of the Tm1 of slow skeletal muscle troponin T. Patient two carried the heterozygous nonsense variant c.334G>T in exon 9 leading to a predicted stop codon p.(Glu112*) in Tm1. Through muscle transcript analysis, a second heterozygous variant was identified in this patient in intron 4, c.74–67C>A, activating a cryptic splice-accepting site and predicted to lead to intron retention. Patient 3 revealed a very early homozygous stop codon p.(Glu6*). Tm1, tropomyosin binding site 1 domain; TNNT1, troponin T type 1.
Analysis of the skeletal muscle transcripts of TNNT1 showed skipping of exons 8 and 9 (figure 4). Western blot analysis revealed the total absence of the troponin protein. No smaller band corresponding to the truncated protein was visible (figure 5). The set of clinical and anatomopathological data was compatible with an implication of this mutation in the patient’s pathology.
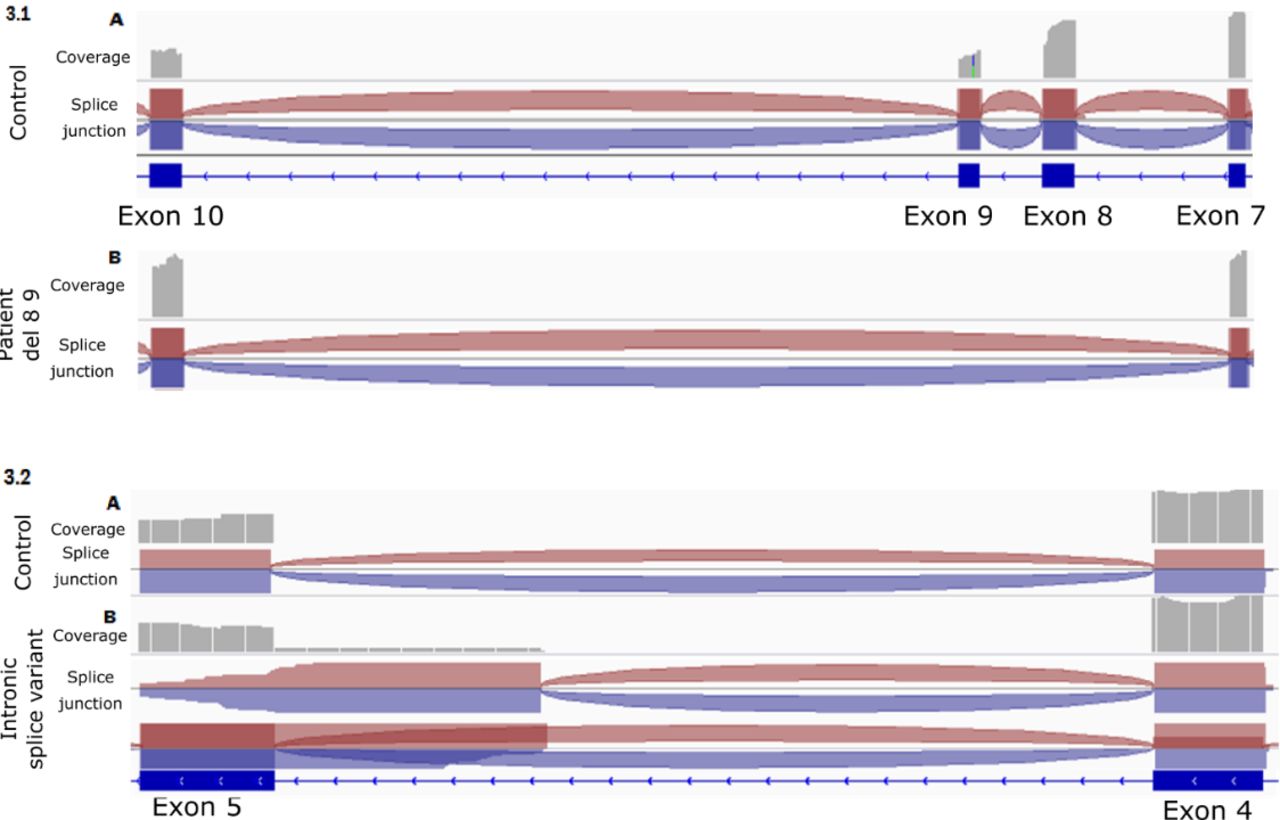
Transcript analyses from muscle biopsies. Visualisation on Integrative Genomics Viewer . In grey are represented the coverage of each sequence. The red and blue crescent represent the splice junction. 3.1. Transcript patterns from exons 7 to 10 from a healthy control (A) and patient 1 (B) samples. The complete skip of exons 8 and 9 observed in patient 1 is represented, compared with the normal splicing of exons 7–10 in the healthy control . 3.2.Transcript patterns from exon 4 to 5 from a healthy control (A) and patient 2 (B) samples. Two transcripts were identified in patient 2: one normal splice pattern between exon 4 and 5 (similar to the healthy control) and another transcript with retention of 65 nucleotides of intron 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Western blot analysis of sTNT. Muscular biopsy homogenates were realised for the three patients (patients 1–3) and an age relative control biopsy (CTRL). An antitubulin antibody was used to control the protein amount loading. An anti sTNT was used to evaluate the quantity of sTNT. Compared with the control, the three patients have nearly null expression of sTNT. The very faint bands detected in patients’ biopsy most probably correspond to non-specific reactivity of the antibodies but could also reflect cross-reactivity with other TNNT isoforms. They represent less than 0.5% of the TNNT1 amount in the control biopsy (TNNT1/tubulin amount fixed to 100% in control). sTNT, skeletal muscle-specific troponin T; TNNT1, troponin T type 1.
Patient 2
This 14-month-old child was born at term to non-consanguineous parents. She was eutrophic, showed a good adaptation to extrauterine life and presented with a congenital torticollis. Axial and peripheral hypotonia was observed only at 3 months, predominant in the pelvic and scapular belts and associated with tongue fasciculations and an abolition of osteotendinous reflexes. At 5 months, a rapid onset of dorsal kyphosis was noted, associated with bilateral limitation of hip and knee amplitudes and pectus carinatum at about 14 months. She developed better motor skills in the forearms and legs. There was no cardiomyopathy. Language developments and interactions were normal for her age. Death occurred at 16 months.
The brain MRI was normal, as were the CPK values. Electroneuromyography revealed a myogenic pattern, but some sequences have neurogenic potentials on the anterior leg. The quadriceps muscle biopsy performed at 9 months was in favour of NM. Many of the muscle fibres contained clusters of rods distributed over the entire cytoplasm. Numerous annular fibres, irregularly sized fibres and endomysial fibrosis were also observed. No correlation between alterations and fibre type could be made, although most of the small fibres were type 1. Ultrastructural analysis revealed numerous muscle fibres with zones of disorganised structure and rods distributed throughout the fibre. Autophagic elements were also observed in several muscle fibres (figure 1). Molecular NGS analysis detected a heterozygous nonsense mutation (c.334G>T; p. (Glu112*)) in exon 9 of the TNNT1 gene, leading to a stop codon on this allele. In addition, the study of TNNT1 transcripts from muscle biopsy revealed partial intronic retention of the last 65 bases of intron 4 of TNNT1, predicted to interrupt the reading frame (figure 4). Genomic sequencing of intron 4 then revealed a variant c.74–67C>A predicted to activate a splice-accepting cryptic site (Human Splicing Finder score of 92.21 and MaxEntScan 12.24), in agreement with the transcript pattern (figure 3). The western blot analysis further revealed the absence of the sTNT protein in the presence of those two mutations, confirming TNNT1 gene involvement in the patient’s pathology (figure 5).
Patient 3
This 6-month-old child was born at 37 weeks and had no difficulty in adapting to extrauterine life. The parents were of North African origin and consanguineous. Their older daughter is in good health. This patient presented with neonatal hypotonia, predominantly axial and notably facial, with the progressive onset of respiratory failure and laryngomalacia associated with swallowing disorders and nutritional deficiency. Hyporeflexia was also associated. Alertness and social interactions were normal for this age. The patient died at 6 months from a respiratory infection. The results of CPK testing, brain MRI, ocular fundus examination and echocardiography were normal. Right diaphragmatic palsy was observed. The muscle biopsy performed at 6 months revealed an NM, with a disproportion in the fibre size: type I fibres were consistently smaller than type II fibres. The nemaline rods in the type I fibres were visualised with Gomori trichrome staining and electron microscopy and showed a homogeneous typical grid pattern. Histoenzymological staining revealed no endomysial fibrosis or fibre grouping. The proportion between the fibre types was normal (figure 1). Sanger sequencing identified a homozygous nonsense mutation in the TNNT1 gene (c.16G>T, p. (Glu6*), leading to a stop codon on both alleles (figure 3). The predicted absence of TNT in the skeletal muscle was confirmed by western blot analysis (figure 5).
Discussion
All patients with NM who present with recessive mutations in TNNT1 have a similar clinical phenotype. The major features are generalised hypotonia with delayed motor development contrasting with conserved fine motor skills and more or less significant joint retraction that may affect the hips, knees and shoulders. Thoracic dystrophies like progressive pectus carinatum indicate progressive respiratory failure that requires invasive or non-invasive ventilatory support, as reported in a Dutch patient and three Palestinian patients.2 9 The semiology of our patients was consistent with these reports, summarised in table 2.
Clinical and genotypic data of patients with TNNT1 pathogenic variants in our series and from previous publications
The presence of tremor noted in the first month of life of patient 1 has been described in all the patients reported in the literature, with the exception of the Dutch patients, who presented either a homozygous mutation at the donor splice site of exon 8 of the TNNT1 gene causing exon 8 skipping in transcripts, or the same heterozygous mutation associated with a deletion of exon 14. Patient 2 presented tongue fasciculations with a myogenic and discreetly neurogenic electromyography: in the absence of dedicated electrophysiological exploration, it is, however, difficult to differentiate between neurogenic damage as already described in patients with TNNT1 mutations electrophysiologically and anapathomopathologically by Abdulhaq et al 2 or tremor also related to the pathology.
Kyphoscoliosis was present in two of four patients, as in the Dutch patients, in the Hispanic patient with a nonsense mutation of exon 9 of the TNNT1 gene and in the Palestinian families. This observation particularly reflects the major axial hypotonia of these patients. In five Palestinian patients, foetal hypomobility and spinal stiffness were also reported, which were less observed in the Amish patients. An interesting observation in one of the Dutch patients was the development of a diaphragmatic hernia at 2 years of life. It was described as secondary to atrophy of the diaphragm. This was the only patient with this complication. Patient 3 presented an elevation of the right diaphragmatic dome, indicating possible diaphragmatic paralysis. The pathophysiology remains unexplained, and this symptom is suggestive of a spinal muscular atrophy with respiratory distress type 1.14 Finally, feeding difficulties and slow weight gain were frequently found, requiring the placement of a gastrostomy tube. Respiratory failure led to the early death of patients in the absence of ventilatory support: before 2 years of age in the Amish population4 and up to 11 years old in the Palestinian population.2 Our three patients died early, despite non-invasive ventilatory support.
Some of the members of a family of Ashkenazi Jewish origin were found to carry an autosomal dominant mutation, giving rise to a less severe phenotype that emerged between 5 and 10 years of age. The phenotype included hypomimia and slowly progressing proximal muscle fatigability, which in some cases progressed to osteoarticular deformities, such as pectus carinatum and/or kyphoscoliosis. Intellectual, cardiac and respiratory functions were not impaired. Survival was not affected. These cases are less severe because of the heterozygosity of the mutation.11
NM is characterised by the presence of rod-shaped inclusions in the skeletal muscle fibres.3 According to the muscle biopsies of our three patients, the rods were more often located in type I fibres. In patient 2, however, the distribution was more homogeneous throughout the cytoplasm and associated with areas of sarcomeric disorganisation, located more often in the centre of the fibre instead of its periphery. These features were emphasised by Johnston et al.4 In addition, as frequently observed in the series of Abdulhaq et al,2 our three patients showed a disproportion in fibre size, with type I fibres consistently smaller than type II fibres, and early endomysial fibrosis. The proportion of type I and type II fibres was preserved, as in previous series.2 4 For patient 1, the rods were not visible with Gomori trichrome staining but only with electron microscopy. This should be considered in relation to the findings in the series of Abdulhaq et al,2 who reported that the rods were often rare and difficult to detect, with no rods observed in three of seven muscle biopsies. It should be emphasised that the ultrastructural aspect of the rods that we observed was quite typical, with homogeneous and electron-dense Z-disk-like lattice structures in continuity with thin filaments, as in all reported cases of NM associated with alterations of the TNNT1 gene.2 4 In particular, no amorphous material was observed within the rod structure, as was described in NM associated with the LMOD3 gene.15
The pathophysiology of NM linked to mutations of the TNNT1 gene is not fully understood, but the absence of slow TNT leads to size defect in both slow and fast fibres,16 which may be the cornerstone of the disease. Functional studies performed in recessive cases due to TNNT1 truncating mutations at Ser108 and Leu203 and to the RNA exon 8 internal deletion revealed a unique impact on the Tm-binding affinity of TNT.17 In patient 1, deleted for exons 8 and 9, as the deleted TNNT1 transcripts were detected, the absence of the protein with western blotting suggests a mechanism of post-translational degradation of TNT, as previously described for the Glu180 nonsense mutation.18 19 This degradation could be the consequence of a complete lack of interaction with Tm. In patient 2, the nonsense mutation at Glu112 in exon 9 of the TNNT1 gene, associated in the other allele with a partial intron 4 retention disrupting the open reading frame, resulted in the total absence of TNT1. In patient 3, the homozygous mutation induced a very early stop codon at Glu6 of the protein and consequently the absence of protein synthesis, as confirmed by the western blot study.
In conclusion, the three patients reported in this series reinforce the homogeneous character of the phenotype associated with recessive forms of TNNT1 mutations. In addition, functional studies show a complete loss of protein in all three cases, consistent with the recent results obtained on TNNT1 knockout mice.20
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Ethics approval
The study was approved by the ethical guidelines issued by our institutions for clinical studies in compliance with the Helsinki Declaration (MR004 : 2 206 723 v 0).
References
Footnotes
JG, KD and JR contributed equally.
MC and CC contributed equally.
Contributors JG, KD, JR, MC and CC contributed to medical evidence, data collection, article writing and reading. EUC, NBR and AS contributed to medical evidence, article writing and reading. PM, A-CC, MK and RJM contributed to article writing and reading. MD, EB, CI, JB, JF, EL, AM, HP, IM and CT contributed to data collection and scientific expertise.
Funding This work was supported by the AT3C Association. We are indebted to Pr Jian-Ping Jin for providing the skeletal muscle-specific troponin T antibody.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.