Article Text

Abstract
Background LMNA-related muscular dystrophy is caused by mutations in LMNA gene. We aimed to identify genetic variations and clinical features in a large cohort of Chinese patients with LMNA mutations in an attempt to establish genotype-phenotype correlation.
Methods The clinical presentations of patients with LMNA-related muscular dystrophy were recorded using retrospective and prospective cohort study. LMNA mutation analysis was performed by Sanger sequencing or next-generation sequencing. Mosaicism was detected by personal genome machine amplicon deep sequencing for mosaicism.
Results Eighty-four patients were identified to harbour LMNA mutations. Forty-one of those were diagnosed with LMNA-related congenital muscular dystrophy (L-CMD), 32 with Emery-Dreifuss muscular dystrophy (EDMD) and 11 with limb-girdle muscular dystrophy type 1B (LGMD1B). We identified 21 novel and 29 known LMNA mutations. Two frequent mutations were identified: c.745C>T and c.1357C>T. A correlation between the location of mutation and the clinical phenotype was observed: mutations affecting the head and coil 2A domains mainly occurred in L-CMD, while the coil 2B and Ig-like domains mainly related to EDMD and LGMD1B. We found somatic mosaicism in one parent of four probands. Muscle biopsies revealed 11 of 20 biopsied L-CMD exhibited inflammatory changes, and muscle cell ultrastructure showed abnormal nuclear morphology.
Conclusions Our detailed clinical and genetic analysis of 84 patients with LMNA-related muscular dystrophy expands clinical spectrum and broadens genetic variations caused by LMNA mutations. We identified 21 novel and 29 known LMNA mutations and found two frequent mutations. A correlation between the location of mutation and the clinical severity was observed. Preliminary data suggested that low-dose corticosteroid treatment may be effective.
- clinical genetics
- muscle disease
- neuromuscular disease
Data availability statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The LMNA gene (OMIM 150330) is located on chromosome 1 in q21.1–21.3, encoding the nuclear envelope intermediate filament proteins, lamin A/C.1 These proteins play an important role in several cellular functions. Mutations in LMNA and other genes of the nuclear envelope can lead to a series of diverse diseases called laminopathies. Laminopathies involve in multiple organ systems including skeletal muscle, heart, fat, peripheral nerves, bone and skin. They are associated with a wide range of disease phenotypes ranging from neuromuscular, cardiac and metabolic disorders to premature ageing syndrome.2 3 Three types of muscular dystrophies are associated with LMNA mutations: the LMNA-related congenital muscular dystrophy (L-CMD, OMIM 613205), the autosomal-dominant (AD) Emery-Dreifuss muscular dystrophy (EDMD2, OMIM 181350) and the autosomal-recessive (AR) EDMD (EDMD3, OMIM 616516).4 Limb-girdle muscular dystrophy type 1B (LGMD1B) is now classified as EDMD2 according to the new classification.5 The clinical severity of LMNA-related muscular dystrophies differs greatly, exhibiting severe to mild muscle weakness and muscle atrophy, contractures and spinal deformities. Cardiac involvement shows a high penetrance, characterised by conduction defects and dilated cardiomyopathy.6 Severe arrhythmia and respiratory failure were common causes of death in the neuromuscular phenotype. Muscle biopsy samples from patients with early-onset L-CMD showed marked inflammatory changes, with a large number of inflammatory cells infiltration.7
Our recent nationwide multicentre study of congenital muscular dystrophy (CMD) was the first to identify L-CMD as the fourth most common subtype of CMD in the Chinese population.8 However, the genotype-phenotype relationship in LMNA-related muscular dystrophy is still unclear. Here, we conducted clinical and genetic analysis of 84 patients with LMNA-related muscular dystrophy from five centres in China and attempted to use data collected from this large cohort to identify genotype-phenotype correlations.
Methods
Patient enrolment
We initially chose patients from 34 tertiary academic hospitals in China for this study but only five centres have diagnosed LMNA-related muscular dystrophies, finally enrolled 101 patients between 2007 and 2019. The inclusion criteria were as we reported before.8 Eighty-four of those were found to have harboured LMNA mutations in the long-term follow-up cohort study. These patients were clinically diagnosed with L-CMD, EDMD and LGMD1B. Patients who had progressive generalised hypotonia or muscle weakness affecting mainly axial muscles or delayed motor milestones presenting within 2 years of life, with or without multiple joint contractures and spinal deformity, were classified as L-CMD. Patients with normal or mildly delayed motor milestones, childhood onset contractures of elbow, posterior cervical and ankle joints, scapulohumeroperoneal weakness and/or cardiac arrhythmias, conduction defects and cardiomyopathy were diagnosed with EDMD. LGMD1B was diagnosed in cases of prominent pelvic and scapular girdle muscle weakness and absent or late onset mild contractures.3 6 9
Clinical and pathological studies
We performed retrospective review of medical records and continued with a prospective cohort study. Clinical and laboratory data, including motor development, family histories, serum creatine kinase (CK) levels, electrocardiography (ECG), ultrasound cardiography and electromyography (EMG) findings were collected. Muscle biopsies were performed, and frozen sections (6 µm) were processed for routine histological and immunohistochemical staining.
LMNA mutation analysis
Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols. All the exons of LMNA and their flanking intronic regions were amplified by polymerase chain reaction (PCR) and directly sequenced in 21 patients. DNA from another 63 patients was analysed by next-generation sequencing (NGS) after year 2013. The candidate variants detected by NGS were verified by Sanger sequencing in the probands and family members. The pathogenicity of the variants was evaluated by the variant-classification guidelines of the American College of Medical Genetics and Genomics and determined based on population frequency, in silico prediction programmes and reporting in locus-specific databases. cDNA analysis was also performed in the cases of novel splicing variants.7
Pathogenic variants determined to have arisen de novo were analysed according to published protocols.10 We collected parents’ blood DNA from six affected children to determine and quantify mosaicism by amplicon resequencing using a personal genome machine (PGM) called PGM amplicon deep sequencing for mosaicism (PASM).
Statistical analysis
We used a Kaplan-Meier plot and log-rank analysis to calculate the age-related rate of ambulation loss and cardiac involvement. A Cox regression model was used to assess the effects of four factors on ambulation loss and cardiac involvement. χ² test was used to compare the relationship between gene mutation and phenotype. A p value of less than 0.05 was considered statistically significant. SPSS software V.21.0 (IBM-SPSS, Chicago, Illinois, USA) was used for data analysis.
Results
In total, 84 patients were identified between 2007 and 2019 to have both clinical diagnosis of LMNA-related muscular dystrophies and confirmed LMNA gene mutations. Of the 84 patients, 41 were diagnosed with L-CMD, 32 with EDMD and 11 with LGMD1B.
Clinical characteristics
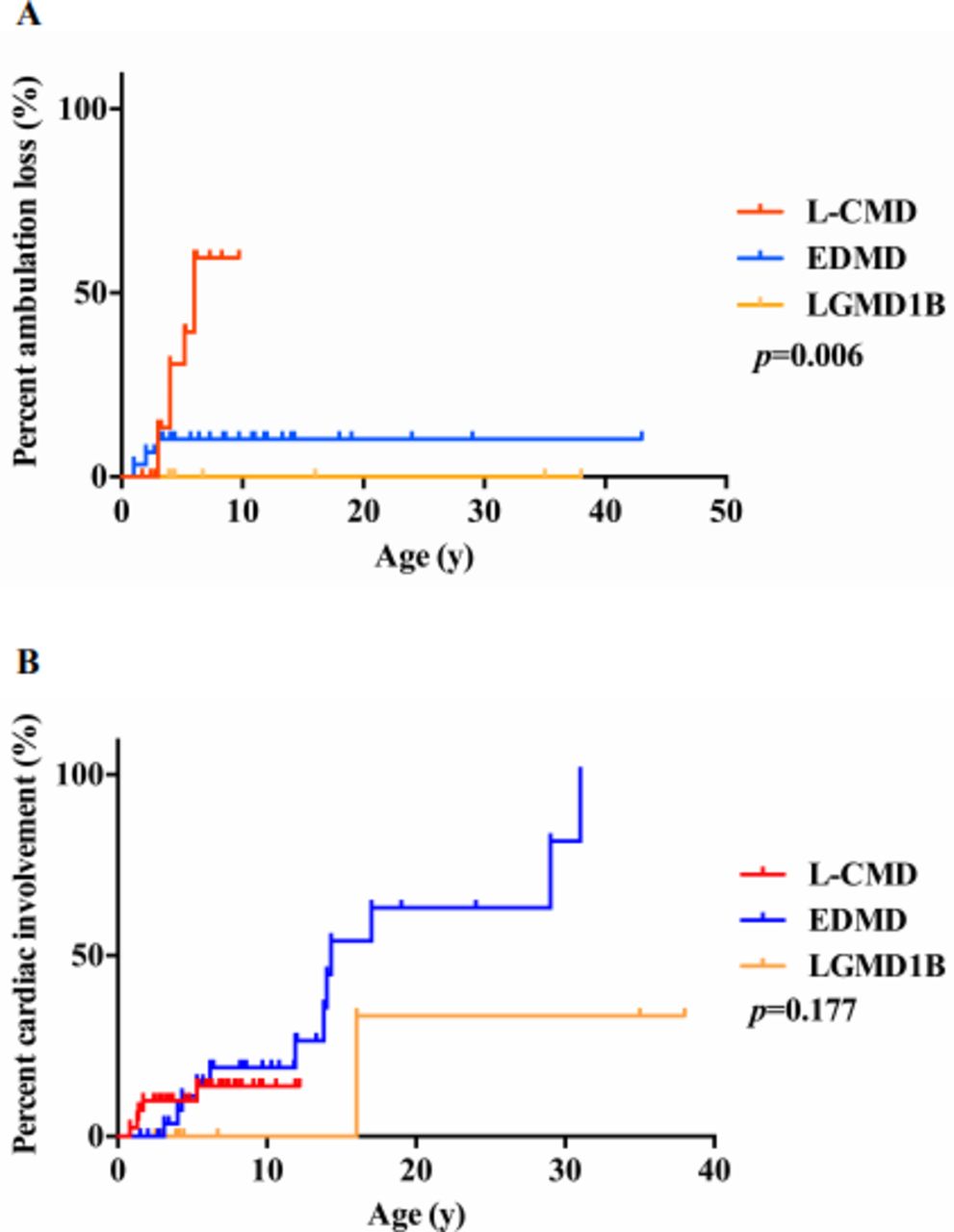
The main clinical characteristics of the 84 patients are summarised in online supplementary table S1. Patients with L-CMD, EDMD and LGMD1B presented with different clinical features in their age of onset, maximal motor function achievement, progression of muscle weakness and orthopaedic deformities. The mean age of onset was 0.3 years for patients with L-CMD, 2.2 years for EDMD and 2.6 years for LGMD1B (table 1, p<0.001). For motor function achievement and regression, patients with L-CMD presented with typical cervical-axial muscle weakness, delayed motor milestones with hypotonia and rapid progression, scapulohumeroperoneal weakness and distal lower extremity weakness. Twenty-eight of 41 patients with L-CMD had typical head drop. Twenty-seven were never able to walk independently (some were able to sit unsupported). Fourteen were able to walk unsupported but with significant gait abnormality and never able to run, jump, ascend or descend stairs. Seven patients had regression with loss of ambulation and became wheelchair dependent at an average age of 4.5 years (between 3 and 6 years). Patients with EDMD exhibited slowly progressive scapuloperoneal muscle weakness. All patients with EDMD could walk independently and all presented with gait impairment and difficulty in running and jumping, ascending and descending stairs and standing up from squatting. Patients with LGMD1B showed relatively mild pelvic and scapular girdle muscle weakness and achieved primary motor milestones on time but exhibited slow walking along with difficulty in ascending stairs and standing up from squatting. In comparisons of ambulation loss, the proportion of patients with L-CMD who achieved ambulation with earlier ambulation loss (median age 6 years) was significantly higher than that of patients with EDMD and LGMD1B (p=0.006, figure 1A and online supplementary table S2). In the area of orthopaedic deformity, patients with L-CMD (lordosis) and EDMD (rigid spine or scoliosis) showed more spinal deformities than patients with LGMD1B (table 1). Spinal deformity occurred earlier in patients with L-CMD (average age 3.1 years) compared with those in patients with EDMD (average age 11.1 years, p<0.001). Joint contractures occurred in 51.2% of L-CMD (average age 4.2 years), 75.0% of EDMD (average age 9.6 years) and only 18.2% of patients with LGMD1B (average age 20.5 years, p<0.001). In patients with L-CMD, the contracture first involved the ankle, followed by the knee, hip and elbow joint. In patients with EDMD, the ankle was the first to be involved, followed by the elbow joint. Two patients have expired so far in this cohort. P1 died of cardiac arrest at the age of 20 and P3 died at the age of 8 years from an unknown cause.
Supplemental material

The clinical features in patients with LMNA-related muscular dystrophy. (A) Comparison of ambulation loss in patients with LMNA-related muscular dystrophy. (B) Comparison of cardiac involvement in patients with LMNA-related muscular dystrophy. EDMD, Emery-Dreifuss muscular dystrophy; L-CMD, LMNA-related congenital muscular dystrophy; LGMD1B, limb-girdle muscular dystrophy type 1B.
Comparison of clinical findings in LMNA-related muscular dystrophy
Cardiac involvement
The EDMD group had a higher risk of cardiac involvement than the L-CMD and LGMD1B groups (figure 1B, table 1 and online supplementary table S2). ECG showed sinus tachycardia in five patients with L-CMD (average age of 2.1 years), four patients with EDMD (average age of 6.9 years) and one patients with LGMD1B (P59; 16 years old), but the cardiac functions and sizes were normal by their echocardiograms. Only one patient with EDMD (P5’s father) showed sinus bradycardia with an average heart rate below 50 beats per minute at the age of 31 years. ECG showed frequent atrial extrasystole in P12 (11 years old) and atrioventricular block in P24 (14 years old). In addition, echocardiogram showed a patent foramen ovale in P47 (3 years old). Notably, P1 was diagnosed with heart failure and pulmonary hypertension at the age of 17. Her ECG showed atrial arrest and a junctional escape rhythm, and the echocardiogram indicated enlargement of the right heart and decreased systolic and diastolic function of the whole heart. She died of cardiac arrest at the age of 20. All patients with LGMD1B except P59 had normal cardiac function.
Respiratory function involvement
Only eleven patients (four L-CMD and seven EDMD patients) underwent a pulmonary function or polysomnography (PSG) test. The predicted forced vital capacity (FVC) was 57% and 57.6% for two patients with L-CMD at 9 and 6 years, respectively. The predicted FVC was 84.6%, 83.9%, 90.0%, 74.2% and 96.8% for five patients with EDMD at 11, 12, 11, 12 and 9 years old. The median FVC at last review of the patients with L-CMD was 57.3%, while that of the patients with EDMD was 84.6% (p=0.133). PSG showed that the minimum oxygen saturation (SpO2) was 56.0%, 76.0% and 66.0% for three patients with L-CMD at aged 9, 9 and 6 years; and 87.0% and 96.0% for two patients with EDMD aged 11 and 9 years. The median minimum SpO2 at last review was 66.0% in patients with L-CMD and 91.5% in patients with EDMD (p=0.200).
Tests of CK and EMG
CK levels were mildly to moderately increased in all 84 patients (see table 1). There was no significant difference among the three clinical subgroups (p=0.797). In the L-CMD group, EMG showed myogenic changes in 19/30 patients, neurogenic changes in 3/30 patients and normal muscle activity in 8/30 patients. Among patients with EDMD, EMG detected myogenic changes in 23/28 and normal activity in 5/28. Six patients with LGMD1B showed myogenic changes.
Muscle pathology
Muscle biopsies were performed on 42 patients (20 L-CMD, 16 EDMD and 6 LGMD1B). Of the patients with L-CMD, 18 revealed dystrophic changes (increased variation in fibre size, necrotic and regenerative fibres and proliferation of connective tissue) and 2 with myopathic changes (atrophic fibres, mild increase in internal nuclei and regenerative fibres). Of the patients with EDMD, 13 showed dystrophic changes and 3 with myopathic changes. In the LGMD1B group, all six patients showed dystrophic changes. Eleven biopsy specimens from 20 patients with L-CMD and 2 of the 16 patients with EDMD exhibited inflammatory changes, with a large number of lymphocyte or mononuclear cell infiltration. Inflammatory cellular infiltration was diffusely seen in the whole muscle specimen, including necrotic fibres, non-necrotic fibres, endomysium and the perivascular region of the perimysium. We previously reported the muscle cell ultrastructures of 10 patients, which showed abnormal nuclear morphology, such as heterochromatin condensation, focal loss of the nuclear membrane and nuclear inclusion bodies.7
Genetic analysis
All 84 patients had mutations in LMNA gene (figure 2A), which are listed in online supplementary table S3. In total, four types of mutations were identified: missense (83.3%), small in-frame deletions (11.9%), splicing (2.4%) and frameshift (2.4%) (figure 2B). Missense was most common mutation in all three groups (table 2). We identified 21 novel and 29 known (10 of which were reported in our previous work7) LMNA gene mutations (figure 2C). All LMNA mutations arose de novo except for those in P5, P48 and P66, who inherited the mutation from their affected parents. No recessive (homozygous or compound heterozygous) LMNA mutations were found in our cohort.
Association among type of LMNA mutations

The genetic features in patients with LMNA-related muscular dystrophy. (A) The phenotype of the 84 patients with LMNA mutations. (B) Distribution of types of LMNA mutations. (C) Schematic of the mutations identified in LMNA . Mutations in upper were novel in our study, while the lower mutations were reported. EDMD, Emery-Dreifuss muscular dystrophy; L-CMD, LMNA-related congenital muscular dystrophy; LGMD1B, limb-girdle muscular dystrophy type 1B.
We also found somatic mosaic mutations in a parent of four probands by PASM, indicating decreased proportions of the mutant allele (7.0%, 3.4%, 9.2% and 18.1%) in the parent (figure 3A), who had no symptoms of LMNA-related muscular dystrophy. Notably, the asymptomatic mother of P30 who was proven to have somatic mosaicism underwent a prenatal diagnosis and found that the fetus carried the same mutation as the proband, which means the mother might have had germline mosaicism (figure 3B).

{kind=link}
{kind=link}
{kind=link}
Mosaicism of mutant alleles determined by PASM. (A) Fractions of mutant alleles in blood samples from six families, identified by PASM. (B) P30’s mother in family 4 underwent prenatal diagnosis, and the fetus carried the same mutation as the proband. FMA, fraction of mutant allele; F, father; M, mother; PASM, personal genome machine amplicon deep sequencing for mosaicism; P, proband.
Phenotype-genotype correlation
We identified two frequent mutations in our cohort: c.745C>T (p.R249W) occurred in 12/41 patients with L-CMD and c.1357C>T (p.R453W) occurred in 5/32 patients with EDMD. Five mutations in L-CMD—c.143G>C (p.R48P), c.745C>T (p.R249W), c.1117A>G (p.I373V), c.1325T>G (p.V442G) and c.1489–14_1489-7del (p.I497_E536del)—and two mutations in EDMD—c.746G>A (p.R249Q) and c.1124C>G (p.A375G)—were associated with inflammatory changes in muscle biopsies.
There appeared to be some correlation between the distribution of mutation sites and certain clinical phenotype. The mutations in the 84 patients were distributed mainly in exons 1 (21.4%), 4 (21.4%), 6 (20.2%) and 7 (15.5%), affecting the head (14.3%), coil 2A (17.9%), coil 2B (21.4%) and Ig-like fold (25.0%) domains (see figure 2 and table 3). Most missense mutations were clustered in exons 1, 4 and 6. The small in-frame deletions were present mainly in exon 1. In patients with L-CMD, mutations were present mainly in exons 1, 4 and 6, affecting the head and coil 2A domains. In patients with EDMD, mutations mainly presented in exons 1, 4, 5, 6, 7 and 9, and in patients with LGMD1B, mutations were showed mainly in exons 6, 7 and 9. The latter two groups mostly affected the coil 2B and Ig-like domains (p=0.0026).
Association between lamin A/C domains and phenotypes
Corticosteroid treatment
Four patients (P10, P13, P16 and P31) were treated with corticosteroid in an attempt to delay disease progression. Muscle biopsies showed obvious inflammatory changes in P10 (L-CMD) and P16 (EDMD), and they received prednisone treatment (0.5 mg/kg/day) started at the age of 1.3y and 7.4y, respectively. There was slight improvement in muscle strength after treatment in P10, and she could lift her upper and lower limbs when lying but were still not able to sit (7 years old now), with poor head control and muscle atrophy. P16 was in stable condition without obvious benefit and stopped the corticosteroid treatment after 3 months. P13 (L-CMD)’s muscle biopsy exhibited no inflammatory changes. He received prednisone (0.7 mg/kg/d) at the age of 3.2y and had a remarkable improvement. He could stand up rapidly from squatting and fell less. P31 (L-CMD) did not undergo muscle biopsy and was treated with methylprednisolone (1.2 mg/kg/d) at the age of 3.4y with muscle strength improvement and could sit unsupported. All the patients were evaluated by the modified MRC (medical research council) muscle strength grading standard to monitor the change of muscle strength and the objective evaluation of motor function (data not shown).
Discussion
In this study, a large cohort of 84 Chinese patients from 81 families were clinically and genetically analysed in detail to identify genotype-phenotype correlations. In this cohort, we characterized three clinical phenotypes associated with LMNA gene mutations: early-onset L-CMD (48.8%), EDMD (38.1%) and LGMD1B (13.1%). The proportions of each phenotype are somewhat different from the Italian cohort of LMNA-associated myopathies with, 23% L-CMD, 22% EDMD and 47% LGMD1B,3 mostly due to the majority patients are children in our cohort. Consistent with the literature, patients with L-CMD in our study exhibited the most severe clinical manifestations and rapid progression with early onset. Head drop and lordosis are typical due to cervical-axial muscle weakness.11 Patients with EDMD presented with relatively late onset and the clinical triad of contracture of the elbows, Achilles tendons and neck extensors; slowly progressive muscle wasting and scapuloperoneal weakness and abnormal cardiac conduction.12 Although the severity of disease was different in these two phenotypes (L-CMD and EDMD), the distribution of muscle involvement seems to be similar. LGMD1B begins in childhood and progresses to adulthood; elbow contracture and restrictive neck flexion are not evident at the early stage, can walk unaided and muscle weakness progresses more slowly. LGMD1B was reclassified as EDMD2 by the LGMD workshop study group due to the high risk of cardiac arrhythmia.5 However, almost no patients with LGMD1B had evidence of cardiac involvement in our cohort. This warrants longer follow-up.
The long-term prognosis of LMNA-related muscular dystrophy is poor, with a high rate of disability and mortality due to progressive motor impairment, contractures, spinal deformities, respiratory insufficiency, cardiomyopathy and various arrhythmias. The common causes of death in LMNA-related muscular dystrophies are severe cardiac arrhythmias and respiratory failure.13 A meta-analysis noted that 92% of patients with LMNA gene mutations have cardiac dysrhythmias after the age of 30 years, and the incidence rate of sudden death in patients with this neuromuscular phenotype is 42%.14 In our study, 13.1% of patients had cardiac involvement before the age of 31 years in L-CMD but one patient with EDMD died suddenly at the age of 20. Therefore, despite severe cardiac involvements were observed less frequently in L-CMD than in EDMD, we propose that regular cardiac monitoring be done in both types of patients. The prevalence of inappropriate sinus tachycardia was 1.16% in a random middle-aged population of Finland.15 Ten patients (11.9%, 10/84) showed sinus tachycardia in our cohort, which were higher than the prevalence in general population reported. And, this needs to be investigated whether sinus tachycardia is pathological compared with the general Chinese population in different ages (data not available). In addition, sinus tachycardia caused by normal compensatory response, such as fever, crying, exercise or emotional tension, should be excluded. Respiratory involvement is the second most common cause of death due to respiratory muscle weakness, recurrent respiratory infection, spinal deformations, contractures and poor nutritional status.7 In our study, four patients with L-CMD had relatively poor lung function, nighttime hypoventilation and hypoxia that were more severe than in patients with EDMD. Therefore, it is important to regularly perform pulmonary function tests and PSG and to use non-invasive ventilation if necessary.
All mutations identified in our study were de novo dominant (96.4%) except for three inherited dominant heterozygous mutations, and no recessive LMNA mutation was found in our cohort. AR LMNA-related muscular dystrophies are rare, and only few AR-EDMD (EDMD3) were reported caused by homozygous LMNA mutations.16–18 In our opinion, old classification of L-CMD, EDMD and LGMD1B may better describe the disease course and inheritance pattern.
The mutations in the 84 patients in our cohort were distributed mainly in exons 1, 4, 6 and 7 (totally 78.5%), affecting the head, coil 1A, coil 2A, coil 2B and Ig-like fold domains. A correlation between the location of mutation and the severity of the disease was observed in our cohort. The mutations in the group with relatively severe L-CMD mainly affected the head and coil 2A domains, while those in the EDMD and LGMD1B groups affected the coil 2B and Ig-like domains. The coil 1A, coil 2A, coil 2B and Ig-like fold domains of lamin A/C are related to dimerization, structural stability, chromatin binding and interacting protein binding. Abnormalities in these domains affect specific lamin A/C functions.7 14 Mutation c.745C>T and mutation c.1357C>T can be considered as frequent mutations in our cohort. Mutations c.745C>T, c.1072G>A and c.1357C>T have also been reported to be common mutations in other studies.19 Mutations c.94_96delAAG and c.116A>G have been found in patients with both EDMD and L-CMD,19–21 suggesting genetic heterogeneity. At the same site, the phenotypic differences caused by mutations may be related to the different changes in protein structure and function caused by different amino acid substitutions, such as for the mutations c.745C>T (related to L-CMD) and c.746G>A (related to EDMD or L-CMD) in our study. Three mutations (c.905T>C, c.1072G>A and c.1489–14_1489-7del) showed different clinical phenotypes from the previously reported cases. Our patient P44 with c.905T>C had EDMD, while the reported patients had L-CMD.15 Three of our patients with c.1072G>A were diagnosed with both EDMD and L-CMD, but this mutation has been reported to be only associated with L-CMD.19 The mutation c.1489–14_1489-7del caused exon 9 skipping. In previous reports, exon 9 deletion has been found to result in dilated cardiomyopathy or LGMD1B in patients. A mouse model with this mutation showed Hutchinson-Gilford progeria syndrome.21–24 However, our patient presented with early-onset L-CMD and exhibited no symptoms of either dilated cardiomyopathy or progeria. Therefore, our current study seems to expand the spectrum of clinical phenotypes caused by the known mutations.
Interestingly, we found the parents of four patients to have somatic mosaicism, and one mother (of P30) was suspected to have germinal mosaicism, similar to a reported consanguineous family in which two children exhibited early-onset L-CMD likely due to paternal germinal mosaicism.25 Mosaicism, which is caused by a postzygotic mutation occurring in somatic cells, the germline or both,26 may be more common than previously thought—especially in de novo mutations—and can increase the difficulty and complexity of clinical diagnosis, genetic counselling and recurrence risk calculation. Mosaicism for dominant mutations in collagen VI-related myopathies has been reported, and germline mosaicism has also been identified in other genes, such as DMD, ACTA1, PMP22, MPZ and BAG3.27 Consequently, molecular genetic testing, such as quantitative analysis of mosaicism in various tissues, should be performed to assess the mosaicism ratios of mutant alleles in unaffected parents of children with LMNA-related muscular dystrophy, for whom accurate genetic counselling and prenatal diagnosis are necessary.
In our cohort, some patients with L-CMD/EDMD and early onset muscle weakness exhibited inflammatory changes in muscle biopsies. Inflammatory myopathy may be related to the age of onset and course of disease. It is usually observed in patients with onset before 2 years of age.28 Infiltrated inflammatory cells were CD4-positive, CD8-positive or CD20-positive lymphocytes,26 CD68-TLR7 double-positive macrophages, and some myeloid CD11c positive dendritic cell and rare CD138 positive plasma cells were also found due to previous study.29 The presence of large numbers of infiltrating inflammatory cells in muscle biopsies suggests that an inflammatory mechanism is involved in the pathogenesis of LMNA-related muscular dystrophy. Steroid treatment is effective in some patients with LMNA-related muscular dystrophy in our cohort and also in previous reports.28 However, due to the strong subjectivity of MRC, it is not suitable to be used as an observation index in patients who received steroids treatment. Hammersmith functional movement screen and motor function measure are suitable to evaluate the motor function of patients and will be used in the future. The pathogenetic mechanism of the inflammatory response in LMNA-related muscular dystrophy remains unknown and needs further investigation. Other patients with early onset in our study did not exhibit inflammatory changes, possibly suggesting different pathogeneses. To date, many studies have addressed LMNA-related dilated cardiomyopathy and accelerated ageing, but the pathogenesis of LMNA-related muscular dystrophy is still unclear. In a study of LMNA-related dilated cardiomyopathy, RNA sequencing of wild-type and D300N mutant transgenic mice predicted activation of E2F, the DNA damage response, TP53, NF-κB and the TGF β pathway.30 These findings suggested that cell death, cell cycle regulation and inflammation are involved in the pathogenesis of dilated cardiomyopathy. In addition, Lmna-/- mouse embryonic fibroblasts have been found to have significant nuclear malformations that exhibit increased nuclear fragility to mechanical stress.2 It was reported that everolimus can ameliorate fibroblast nucleus defects in patients with laminopathies.31 Therefore, these studies provide some scientific rationale for the further research of pathogenesis of LMNA-related muscular dystrophy.
In summary, we provide detailed clinical descriptions and genetic analysis of 84 patients with LMNA-related muscular dystrophy. Our current study expands the spectrum of clinical phenotype and genotype caused by LMNA mutations. We identified 21 novel and 29 known LMNA mutations. A correlation between the location of mutation and the severity of the disease was observed: the head and coil 2A domains mainly related to L-CMD, while the coil 2B and Ig-like domains mainly related to EDMD and LGMD1B. We found two frequent mutations in our cohort: c.745C>T occurred in 12 patients with L-CMD and c.1357C>T in 5 patients with EDMD. We also found somatic mosaic variant in one parent of four probands, increasing the difficulty and complexity of clinical diagnosis and genetic counselling. The histopathological findings and our preliminary clinical data suggested that low-dose corticosteroid treatment may be effective. Our research provides a pathway towards future pathogenetic studies and possible treatment strategies for LMNA-related muscular dystrophy.
Data availability statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Ethics approval
The study was approved by the Ethics Committee of Peking University First Hospital (No. 2015[916], Beijing, China). Informed written consent was obtained from all patients and/or their legal guardians.
Acknowledgments
The authors appreciated all the grant supports that enabled this research. The authors thank all the health providers, patients and their families who have participated in this study.
References
Footnotes
Contributors YF and DT conducted the patient follow-up and wrote the first draft of the manuscript. DS, LG and H Yang collected the clinical and laboratory data and followed up on patients. JL took part in drafting the manuscript. XZ and SuW analysed the muscle cell ultrastructure of the patients. ZW, YY, CZ, SH-SC, LW and BM provided patients data as multicentre participants. XC analysed muscle biopsies of the patients and ShW conducted and analysed EMG. H Yan helped analyse the cardiac data of all the patients. CB, XW, HZ and HX took part in study design and critical discussions. HZ and HX conceived the study and participated in its design and coordination and helped to draft the manuscript. All authors participated in drafting and critically revising the article and approved the final manuscript.
Funding This work was supported by the National Key Research and Development Program of China (No. 2016YFC0901505); National Natural Science Foundation of China (No. 81571220); Beijing key laboratory of molecular diagnosis and study on pediatric genetic diseases (No. BZ0317); Peking University Medicine Seed Fund for Interdisciplinary Research supported by the Fundamental Research Funds for the Central Universities (No. BMU2017MX003, BMU2018MX001); Natural Science Foundation of Jiangxi Province (No. 20161BAB215192).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.