Article Text

Abstract
Type 1 diabetes mellitus (T1DM) is defined as an autoimmune disorder and has enormous complexity and heterogeneity. Although its precise pathogenic mechanisms are obscure, this disease is widely acknowledged to be precipitated by environmental factors in individuals with genetic susceptibility. To date, the known susceptibility loci, which have mostly been identified by genome-wide association studies, can explain 80%–85% of the heritability of T1DM. Researchers believe that at least a part of its missing genetic component is caused by undetected rare and low-frequency variants. Most common variants have only small to modest effect sizes, which increases the difficulty of dissecting their functions and restricts their potential clinical application. Intriguingly, many studies have indicated that rare and low-frequency variants have larger effect sizes and play more significant roles in susceptibility to common diseases, including T1DM, than common variants do. Therefore, better recognition of rare and low-frequency variants is beneficial for revealing the genetic architecture of T1DM and for providing new and potent therapeutic targets for this disease. Here, we will discuss existing challenges as well as the great significance of this field and review current knowledge of the contributions of rare and low-frequency variants to T1DM.
- diabetes mellitus
- endocrinology
- genetics
- molecular medicine
- sequence analysis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Currently, type 1 diabetes mellitus (T1DM) is defined as an autoimmune disorder classically characterised by pancreatic islet beta-cell destruction triggered by autoreactive T cells, resulting in subsequent severe insulin deficiency and lifelong reliance on exogenous insulin.1 2 This autoimmune diabetes accounts for 5%–19% of diabetes and represents the main form of diabetes in children and adolescents.3 Its incidence is increasing worldwide at a rate of 2%–5% per year.4 This rising incidence and multiple severe diabetic complications lead to increased mortality and morbidity and aggravate the economic burden of the disease. It is accepted that the interplay between genetic factors and environmental precipitators, including ancestry and geographic location, viral and bacterial infections, vitamin D, hygiene and microbiota, leads to specific tissue inflammation, namely, insulitis, insulin-producing cell death and consequent clinical disease.5–9
The genetic component of T1DM can be demonstrated by the fact that siblings and offspring of patients with T1DM have a higher risk than the general population, and disease concordance in identical twins is higher than that in dizygotic twins.10 11 Over the past few years, genome-wide association study (GWAS), which measures and analyses a million or more DNA sequence variations in known linkage regions in unrelated individuals, have identified at least 58 susceptible loci combined with linkage analysis and candidate gene studies (figure 1).12–14 Most of the identified variants are common (minor allele frequency (MAF) >5%) and have modest effects (OR <1.5), although the effects of susceptibility genes such as human leucocyte antigen (HLA), insulin (INS) and protein tyrosine phosphatase, non-receptor type 22 (PTPN22) are stronger (figure 1).13 The HLA region (OR >6), located on human chromosome 6p21 and identified by linkage analysis, accounts for the largest proportion of T1DM heritability and explains approximately 50% of genetic T1DM risk.15 In addition to HLA, variants within the INS and PTPN22 loci, which were first identified by candidate gene studies, have larger effect sizes (OR >2) than other variants.13 The INS gene on human chromosome 11p15.5 offers the next strongest genetic risk association with T1DM after HLA and accounts for approximately 10% of genetic susceptibility to T1DM.16 It is believed that ‘missing heritability’ can be at least partially elucidated by rare and low-frequency variants (rare variants defined as variants with MAF ≤1% and low-frequency variants defined as variants with MAF=1%–5%), and some findings have indicated that rare variants have larger effect sizes than common variants.17–19 From an evolutionary standpoint, risk variants with higher penetrance are more likely to be rare due to negative selection. Taking an extreme example, monogenic/Mendelian disorders such as autoimmune polyendocrinopathy syndrome type I are caused by rare variants with large effect sizes and high penetrance. Intriguingly, recent and previous studies focusing on the identification of rare and low-frequency variants involved in T1DM have found a handful of such variants, and some of them do have large effect sizes.13 20–23
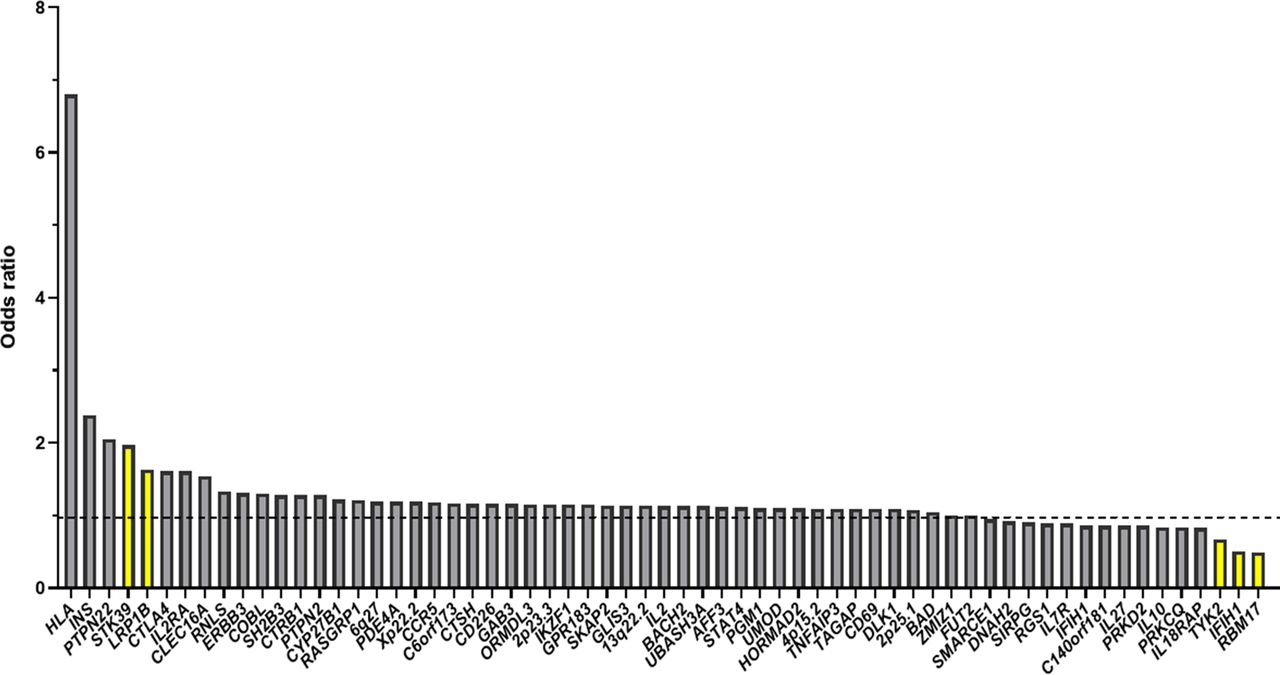
Candidate genes or loci of type 1 diabetes mellitus (T1DM) and their ORs (the yellow bars represent the rare and low-frequency genetic variants of T1DM).76–79
However, some studies suggest that most rare variants have only small or modest effects.24 Therefore, it remains to be seen whether the tendency of rare and low-frequency variants to have large effects is a universal phenomenon. Even though its practical value in clinical medicine may be restricted if the hypothesis that most rare variants have only a small effect is true, there is still intrinsic value in this field. Such studies can lead to the discovery of new candidate genes implicated in disorders or human phenotypes25 and determine causal genes in candidate regions identified by GWAS. Other than understanding better its pathophysiology, new loci could lead to the identification of new biomarkers or represent drug targets for T1DM.
Identifying rare and low-frequency variants
Recently, advances in next-generation DNA sequencing technologies as well as bioinformatic tools and methods to process and analyse the resulting data have enhanced the ability of researchers to find rare variants, and the decreasing cost of these technologies has made it feasible to apply them to related studies (table 1).26 The most comprehensive approach is high-depth whole-genome sequencing (WGS) due to its excellent coverage. However, high costs and multiple computational challenges have restricted its application.21 In addition to WGS with high or low depth, SNP-array genome-wide genotyping and imputation has been used to identify rare variants. Notably, current sequencing depth (especially 30x) of WGS is likely to miss at least some coding variants as compared with whole-exome sequencing (WES, especially >100x).
Technologies and study designs for detecting rare variants
There are some lower-cost alternatives as well. First, a combination of low-depth WGS and imputation is another choice. Imputation is a statistical method that can determine genotypes that are not directly detected by taking advantage of various previously sequenced reference panels. For instance, Martínez-Bueno and Alarcón-Riquelme identified rare variants that were jointly associated with systemic lupus erythematosus (SLE) within 98 SLE candidate genes by applying genome-wide imputation and other techniques.27 Notably, some studies have indicated that the newer imputation panels, such as the recent Haplotype Reference Consortium panel and the combined UK10K and 1000 Genomes projects phase III, provide better quality of imputation for rare variants compared with early panel, such as the UK10K, which underlines the significance and potential of larger reference panels to impute rare variants.28 29 Nevertheless, the power of imputation for identifying rare variants is attenuated because its accuracy decreases with decreasing MAF. Additionally, studies have indicated that the utility of population-specific panels leads to improved imputation accuracy of rare variants.30 Therefore, the utilisation of imputation is relatively limited in non-European populations because of the lack of ethnicity-specific reference cohorts.
Second, using WES finds rare variants within protein-coding regions. Given the reality that only an exceedingly small portion of the human genome is coding sequence and the functions of protein-coding variants are more easily interpreted, WES is considered a cost-effective technique for discovering rare variants. However, an obvious defect is that WES ignores non-coding regions, which account for 98% of the human genome. Moreover, most loci identified by GWAS are located in non-coding regions, and evidence indicates that these regions play critical roles in complex disorders and have significant biological functions.31 32
Third, targeted sequencing investigates a specific part of the genome, including candidate genes identified by previous studies and clinically significant genes. For instance, Rivas et al identified a protein-truncating variant of the gene RNF186 that can exert a protective effect against ulcerative colitis via changed localisation and decreased expression by conducting targeted sequencing in regions previously associated with inflammatory bowel disease; they found that this loss-of-function variant was a promising therapeutic target.33 However, some targeted sequencing studies have failed to detect rare risk variants, indicating the deficiency of this method in discovering rare and low-frequency variants.24 34
In addition, burden tests, which collapse information for multiple variants into a single genetic score and analyse the association between the score and disease characteristic, are a common approach in genomics to potentialise identification of rare variants, because aggregating analysis of variants within a gene can improve the power to detect statistical signals between case and control subjects. For example, a study analysed WES data from 393 patients with idiopathic hypogonadotropic hypogonadism (IHH) against 123 136 control subjects from public sequencing database, and identified a significant burden in TYRO3, a candidate gene implicated in IHH in mouse models.35 However, this gene-based burden testing approach will lose power when effects of variants are not in the same direction or the causal variants only account for a small fraction.36
Traditional genetic studies have focused mostly on DNA sequences collected from unrelated individuals. However, a variety of new study designs have been applied to finding rare variants with the goal of decreasing sample sizes and costs. The common feature of these designs, including extreme phenotype sampling, population isolates and family studies (table 1), is that they improve the power of rare variant testing by selecting a specific population.37–39
Challenges for identifying rare and low-frequency variants
The detection and analysis of rare and low-frequency variants constitute a rising research field, but this field has encountered substantial obstacles and challenges. First, the statistical analysis of rare and low-frequency variants is far more complicated and difficult than the analysis of common variants. For example, because the number of rare variants is greater than the number of common variants, the significance threshold or p value established for GWAS is not appropriate for rare variant association studies.40 The linkage disequilibrium (LD) r2 between two rare variants or a common variant and a rare variant cannot be accurately calculated, and as such it is difficult to define if novel rare variants are independent from known rare or common variants.41 42 A variety of traditional methods used to reduce or eliminate confounding factors and population stratification, such as linear mixed effect models and principal components analysis, are not applicable to the analysis of rare and low-frequency variants because rare variants and the distribution of disease risk are strictly localised. A study indicates that the estimated ancestry scores can be used to control the population stratification if the pool of control is large. Also, off-targeted read might be applied for controlling population stratification in targeted sequencing.43 Moreover, because these variants are rare, the strategy used to analyse common variants, which is based on analysing a single variant at a time, is underpowered to detect rare variants and can do so only if the effect size or sample size is exceedingly large.44 Thus, alternative methods have been developed to analyse the aggregate effect of rare variants.45–47 These methods, such as burden tests, variance component test and exponential combination tests, evaluate association for multiple variants in a gene or a biologically region. Combined analysis of genetic association data with other biological information, such as methylation, gene expression and biological pathways, can also leads to substantial gain In the statistical power of rare variants studies.48–50
Second, it still remains challenging to apply genetic information obtained by rare variants association studies to diagnostic and prognostic medicine because some healthy individuals carry deleterious variants. For example, Flannick et al found that a large portion of the general population carries low-frequency non-synonymous mutations that can change the length or sequence of coding proteins in maturity-onset diabetes of young genes, and these carriers remain normoglycaemic through middle age.51 In addition, Bick et al discovered that rare variants in sarcomere protein genes could boost the risk of adverse cardiovascular events in Framingham Heart Study participants, and more surprisingly, a large number of non-synonymous variants, including nonsense, missense and splice variants, are present in healthy populations.52 Therefore, the functional validation of rare and low-frequency genetic variants is necessary to determine the causality in genotype-phenotype analysis.
Third, many rare and low-frequency variants are geographically localised and population specific, so it is difficult to find suitable replication panels and generate a common population. Nelson et al sequenced 202 drug target genes in coding regions in 14 002 people and found that 95% of observed variants are rare and at least 74% are detected in only one or two individuals.53 Similarly, a study conducted in 2440 individuals of African and European ancestry found that 86% of over 500 000 variants identified are rare, and most are previously unknown.54 Notably, these studies indicate that the vast majority of rare variant allelic spectra are unique to their sample sets and need to be identified by direct resequencing.
Finally, although some detection studies of rare and low-frequency variants, such as WES and data processing software, are relatively standardised, many aspects of this emerging field, including WES capture technologies and even the definition of rare variants, still do not have uniform standards. Therefore, combining data generated from different groups is problematic.
Benefits of identifying rare and low-frequency variants
It has been suggested that rare and low-frequency variants account for a large proportion of the genetic variation in the human genome represented by the 1000 Genomes Project.55 56 Although a substantial number of SNPs have been identified by GWAS, there is still a so-called ‘missing heritability’ phenomenon in complex disorders.57 For instance, GWAS have identified >80 common variants with small effect sizes for T2DM, which can explain only 10% of the total heritability.58 To address this issue, several hypotheses have been proposed, and great technological advances have provided a better understanding of the genetic architecture of common diseases over the past several years. Rare and low-frequency variants can influence both susceptibility to common complex diseases and their phenotypes (table 2).59–62 For example, researchers performed WGS in 1038 pulmonary arterial hypertension (PAH, a rare disorder characterised by occlusion of arterioles in the lung) cases and 6385 control subjects and make the total proportion of cases explained by mutations increased to 23.5% from previously established 19.9% by incorporating novel rare variants and genes identified.63 Also, a study indicated that rare variants of SLC22A12 gene influence urate reabsorption and the heritability explained by these SLC22A12 variants exceeds 10%, indicating that rare functional variants make substantial contribution to the ‘missing heritability’ of serum urate level.64 In fact, a ‘common disease-rare variant model’ that assumes rare variants with high penetrance may be involved in increased complex disease risk has been proposed.59 65 It is obvious that great genetic heterogeneity exists under this model. Intriguingly, in line with this model, some autoimmune diseases, such as T1DM, are extremely heterogeneous.
Rare and low-frequency variants associated with T1DM, T2DM and other autoimmune diseases
Besides rare and low-frequency genetic variants, there are some other hypotheses to explain the ‘missing heritability’.59 For example, empirical and theoretical analyses have indicated that multiple genetic variants with small effects are missed because GWAS are underpowered to capture these variants, therefore, taking into account genetic variants with smaller effects that do not reach significance will contribute to disease susceptibility and phenotype variability. Additionally, structural variants, such as CNV, are poorly studied owing to insufficient coverage on SNP chips.66 The presence of gene-gene (epistasis) and gene-environmental interactions may also contribute to the ‘missing heritability’.67
In addition, the candidate regions identified by GWAS sometimes harbour several different genes. Identifying rare genetic variants is helpful to pinpoint causal genes within the loci identified by GWAS.68 Moreover, the identification of rare and low-frequency variants may result in the identification of new candidate genes.40 For instance, researchers identified a heterozygote truncating mutation within CLCN1 gene by performing WES in patients with statin-associated myopathy and therefore, determined a novel candidate gene of this disease.69 Additionally, it has been suggested that rare variants are likely to have appeared more recently than common variants, leading to reduced LD and making them more easily interpretable than common variants.21
Moreover, early studies have indicated that rare and low-frequency genetic variants may have larger effects on complex disease phenotypes and susceptibility than common variants.70 Therefore, it is helpful to reveal the genetic pathways underlying diseases and to provide clinically actionable targets for personalised medicine. As an example, Roth et al found that rare and low-frequency genetic variants with large phenotypic effects within the proprotein convertase subtilisin/kexin 9 (PCSK9) gene, which encodes products that bind to the low-density lipoprotein (LDL) receptor and increase its degradation, can lower the risk of coronary heart disease (CHD) by reducing the circulating level of LDL cholesterol.71 Based on this research, a fully human monoclonal antibody targeting PCSK9 has been proven to increase LDL receptor recycling and decrease LDL cholesterol level.72 These findings provide a new treatment and prevention strategy for hypercholesterolaemia and CHD and offer inspiration for the transformation of genetic discoveries into clinical practice.
Rare and low-frequency variants and T1DM
Focusing on autoimmune diabetes, fully understanding the genetic factors underlying T1DM is beneficial for revealing its pathophysiology, discovering new drug targets and developing predictive and personalised medicine (figure 2). It is especially vital and valuable because T1DM is extremely complex and heterogeneous. The candidate T1DM loci identified by GWAS sometimes contain several distinct genes, and strong LD makes it difficult to pinpoint the precise causative genes in genomic regions. In addition, the fact that many SNPs reside in non-coding regions or do not have obvious functional effects offers few clues to ascertain the causative genes. However, the discovery of rare and low-frequency disease-associated variants is helpful for T1DM candidate gene identification. The T1DM-associated region on human chromosome 2q24 harbours interferon (IFN) induced with helicase C domain 1 (IFIH1), GCA, FAP and part of KCNH7. The interaction between IFIH1 and double-stranded RNA, a byproduct of viral replication, leads to the secretion of IFNs. While IFIH1 is a plausible susceptibility gene on the basis of its biological function, there is no direct evidence to indicate which of these genes in this locus is responsible for increased T1DM risk. Nejentsev et al resequenced the exons and splice sites of 10 candidate genes in pools of DNA from 480 patients and 480 controls and discovered 4 rare or low-frequency variants (OR=0.51–0.74, MAF <3%) with low LD within IFIH1 that could change the structure or expression of its product, melanoma differentiation-associated protein 5 and protect against T1DM.23 This finding suggests that IFIH1 is the disease-causing gene. Moreover, Ge et al found several rare deleterious variants, including two novel frameshift mutations (ss538819444 and ss37186329) and two missense mutations (rs74163663 and rs56048322) within PTPN22 by deeply sequencing the protein-coding regions of 301 genes in 49 loci previously identified by GWAS in 70 T1DM cases of European ancestry.22 This finding further confirmed that PTPN22 is a T1DM candidate gene on chromosome 1p13.2. Subsequent genotyping in 3609 families with T1DM indicated rs56048322 (MAF=0.87%), which leads to the production of two alternative PTPN22 transcripts and a novel isoform of its encoding protein, LYP, through affecting splicing of PTPN22, was significantly associated with T1DM independent of T1DM-associated common variant rs2476601. Functional analysis showed this isoform of LYP can cause hyporesponsiveness of CD4+ T cell to antigen stimulation in patients with T1DM.

{kind=link}
{kind=link}
The development of type 1 diabetes mellitus (T1DM). T1DM is caused by interplay between genetic and environmental factors, and epigenetics serves as a bridge between the two. To date, >50 candidate loci have been identified by genome-wide association study. The genetic variants within these risk regions can be divided into common variants, low-frequency variants and rare variants according to their different minor allele frequencies. The rare and low-frequency variants are likely to have more practical value in the treatment of T1DM because their ORs are larger than those of common variants. However, as the study of rare and low-frequency variants is an emerging research field, some hypotheses are still controversial and need further investigation. LD, linkage disequilibrium; MAF. minor allele frequency.
Additionally, as mentioned above, most variants that confer T1DM risk are common and have modest effects, limiting the clinical application of their discovery. However, some research has suggested that rare and low-frequency variants might have larger effect sizes than common variants. Theoretically, if a disorder affects reproduction, such as an autoimmune disease with early onset, genetic variants with strong effects will be maintained at a relatively low frequency through negative selection.21 Forgetta et al applied deep imputation of genotyped data in 9358 patients with T1DM and 15 705 controls from European cohorts to identify novel rare and low-frequency variants with large effect sizes on T1DM risk.13 Three novel rare and low-frequency variants, including rs192324744 in LDL receptor-related protein 1B (LRP1B, MAF=1.3%, OR=1.63), rs60587303 in serine threonine kinase 39 (STK39, MAF=0.5%, OR=1.97) and the intergenic variant rs2128344 (MAF=0.55%, OR=2.12), were found and validated by subsequent de novo genotyping.13 Notably, the effects of these SNPs (ORs ≥1.5) are comparable to those of the lead variants in INS and PTPN22. In vitro experiments indicated that STK39 is involved in T cell activation and effector functions and that inhibition of Stk39 can augment the inflammatory response by enhancing interleukin (IL)-2 signalling; therefore, STK39 may be a promising clinical intervention target.13
Besides, previous study through fine mapping of known T1DM susceptible loci has identified a low-frequency variant rs34536443 (MAF=4%, OR=0.67) within tyrosine kinase 2 (TYK2) and a rare variant rs41295121 (MAF=1%, OR=0.49) within RNA binding motif protein 17 (RBM17, in the same locus as IL2RA).20 TYK2, belonging to Janus kinase (JAK) family, is associated with regulation of type I IFN signalling pathway. Some studies have demonstrated that rs34530443 plays protective roles in multiple autoimmune disorders and the underlying mechanisms might lie in the diminishment of IL-12, IL-23 and type I IFN signalling.73 The specific function of rs41295121 in context of autoimmunity and T1DM needs further investigation.
As for some practical issues such as sample sizes and high costs, a study indicated that a well-powered rare variant association study should include discovery sets with at least 25 000 cases and a substantial replication set.44 There are some alternative methods to decrease the sample sizes or costs in the context of T1DM. For example, combined analysis of rare variants within a T1DM-associated gene or region can lead to substantial reduction of required sample sizes. In addition, preferential selection of individuals with extreme phenotype on the basis of known risk factors, including age of disease onset, family history of diabetes and diabetic auto-antibodies, can also improve the association power because rare variants might be enriched among them.74
Overall, among the identified T1DM loci, the candidate genes with rare or low-frequency variants include TYK2, IFIH1, RBM17, PTPN22, STK39 and LRP1B.13 20 22 23 Many unidentified variants may remain to be dissected, because studies focused on other diseases suggest that rare and low-frequency variants account for the majority of all variants.27 75
Conclusion
Driven by advancements in sequencing technologies, there has been great improvement in the identification of rare and low-frequency variants that cause complex human diseases, such as T1DM. The benefits of this field can be stated as follows: (1) characterisation of rare and low-frequency variants may lead to a full understanding of the genetic component of this disorder; (2) detection of rare and low-frequency variants can pinpoint the genes that are actually responsible for increased T1DM risk within the loci identified by GWAS; (3) some new candidate genes for T1DM can be found due to enhanced power to discover rare variants; (4) rare and low-frequency variants are expected to make a significant contribution to human phenotypes and disease susceptibility because some studies indicate the majority of protein-coding variants tend to be evolutionarily recent and rare54; (5) accumulated evidence indicates that rare and low-frequency variants have larger phenotypic effects than common variants, suggesting that they will offer more actionable clinical targets and hold tremendous promise in predictive and personalised medicine.
However, some issues remain to be addressed. First, controversy persists about the importance of rare and low-frequency variants in common diseases. Encouragingly, recent studies have found that some such variants, such as rs60587303 in STK39, indeed have larger effect sizes than common variants in the pathogenesis of T1DM. Second, the candidate genes for T1DM that have rare or low-frequency variants included only TYK2, RBM17, IFIH1, PTPN22, STK39 and LRP1B, which means there may still be many unidentified variants. Moreover, most studies in this field have examined European populations. However, rare and low-frequency variants are geographically localised and population specific. In particular, the heritable background of T1DM varies among different ethnic groups. These facts will limit the practical application of rare and low-frequency variants.
In conclusion, the identification of rare and low-frequency genetic variants will provide new insights into the pathophysiology of T1DM and offer new potential drug targets in the post-GWAS era, despite the many challenges and uncertainties remaining in this field.
References
Footnotes
Contributors HP searched references, wrote the first draft of the paper and revised the text. YX, SL, GH and XL critically revised the text and provided substantial scientific contributions. ZX and ZZ proposed the project and revised the manuscript. All the authors approved the final version of the manuscript.
Funding This work was supported by the National Natural Science Foundation of China (grant numbers 82070813, 81873634, 81400783), the National Key R&D Program of China (grant numbers 2016YFC1305000, 2016YFC1305001, 2018YFC1315603), the Science and Technology Major Project of Hunan Province (grant number 2017SK1020), Hunan Province Natural Science Foundation of China (grant numbers 2018JJ2573, 2020JJ2053).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.