Article Text

Abstract
Background Arthrogryposis multiplex congenita (AMC) is the direct consequence of reduced fetal movements. AMC includes a large spectrum of diseases which result from variants in genes encoding components required for the formation or the function of the neuromuscular system. AMC may also result from central nervous involvement. SCN1A encodes Nav1.1, a critical component of voltage-dependent sodium channels which underlie action potential generation and propagation. Variants of SCN1A are known to be responsible for Dravet syndrome, a severe early-onset epileptic encephalopathy. We report pathogenic heterozygous missense de novo variants in SCN1A in three unrelated individuals with AMC.
Methods Whole-exome sequencing was performed from DNA of the index case of AMC families. Heterozygous missense variants in SCN1A (p.Leu893Phe, p.Ala989Thr, p.Ile236Thr) were identified in three patients. Sanger sequencing confirmed the variants and showed that they occurred de novo.
Results AMC was diagnosed from the second trimester of pregnancy in the three patients. One of them developed drug-resistant epileptic seizures from birth. We showed that SCN1A is expressed in both brain and spinal cord but not in skeletal muscle during human development. The lack of motor denervation as established by electromyographic studies or pathological examination of the spinal cord or skeletal muscle in the affected individuals suggests that AMC is caused by brain involvement.
Conclusion We show for the first time that SCN1A variants are responsible for early-onset motor defect leading to AMC indicating a critical role of SCN1A in prenatal motor development and broadening the phenotypic spectrum of variants in SCN1A.
- human genetics
- genomics
- neuromuscular diseases
- epilepsy
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplementary information. All data relevant to the study are included in the article and are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Arthrogryposis multiplex congenita (AMC) has an overall incidence of 1 in 3000 to 5000.1 2 AMC is the direct consequence of reduced fetal movements which may lead, in addition to AMC, to pterygia, pulmonary hypoplasia, diaphragmatic defect or cleft palate. Some non-genetic factors may cause AMC such as extrinsic limitation of fetal movements, bacterial or viral infections, or maternal autoimmune myasthenia.
Considerable genetic heterogeneity has been reported in AMC. AMC includes a large spectrum of diseases which result from variants in genes encoding components required for the formation or the function of neuromuscular junctions, skeletal muscle, survival of motor neurons, myelination of peripheral nerves or the function of connective tissues.3 AMC may also result from central nervous involvement including channelopathies linked to variants of NALCN (MIM: 611549)4 or CACNA1E (MIM: 601013).5
The identification of the pathogenic variant in AMC is essential to provide information regarding prognosis and comorbidities and informs accurate genetic counselling. Here, we further explored genetic alterations in a group of genetically undiagnosed AMC through whole-exome sequencing (WES) and identified heterozygous de novo missense variants in SCN1A in three unrelated patients. SCN1A (MIM: 182389) encodes a critical component of voltage-dependent sodium channels. Variants of this gene most frequently present as Dravet syndrome (EIEE6, [MIM: 607208]), an early-onset epileptic encephalopathy.6 We present the clinical data for these three patients and describe the phenotypic spectrum associated with this new recurrent genetic disorder.
Methods
The parents of all affected individuals provided written informed consents for pathological examinations and genetic analyses of their affected children or fetuses and themselves in accordance with the ethical standards of our institutional review boards.
Whole-exome sequencing
WES was performed from DNA of the index case in the AMC families using a completed Twist Bioscience Human Core Exome (Consensus CDS) kit for library preparation and exome enrichment (Integragen). Sequencing was performed on a Genome Analyzer Hiseq4000 Illumina instrument in paired-end mode with a read length of 2×75 bp (Integragen). Reads were aligned to the human reference genome sequence (UCSC hg19, NCBI build 37.3) via the BWA program.7 Variants were selected using the SAMtools8 then annotated using Annovar software.9 Variants in coding regions (including non-synonymous and nonsense variants), intron–exon junctions or short coding insertions or deletions were selected when the minor allele frequency was less than 0.005. Prediction of pathogenicity of missense variants was performed using Polyphen-210 and SIFT software.11
Sanger sequencing
Variants identified through WES were validated by Sanger sequencing. The primers used for the detection of the SCN1A c.2679A>C variant are 5′-CTATCCACTCCCCACACAGC-3′ and 5′-GAGACGGTTAGGGCAGATCA-3′; for c.2965G>A: 5′-AGCTACTCCTTTGTGCATCCT-3′ and 5′-CAGTCACAAATTCAGATCACCCA-3′ and for c.707T>C: 5′-CTGCAGCCCAATTAGAGCAA-3′ and 5′-CTTCTCCACTAGCGTTGCAA-3′. PCR amplification was carried out as previously described.12 PCR products were purified then sequenced using the forward or reverse primers (Eurofins Genomics). The obtained DNA sequences were compared with published sequences (BLAST, NCBI). Sanger sequencing was also performed to establish the genotype of each family member and to analyse the segregation of the variant within each family.
Real-time PCR amplification of genomic DNA
Real-time PCR amplification was conducted using genomic DNA on a 7300 Real-Time PCR system (Applied Biosystems) using Power SYBR Green PCR Master Mix (Applied Biosystems).12 Genomic deletion was defined when the ratio of tested DNA to control DNA was equal to or less than 0.5. Real-time PCR amplification of each sample was performed in duplicate using primers within SCN1A exons 1, 10, 14, 15 and 16 (data available on request). Albumin gene was used as internal control (data available on request).
RT-PCR amplification
Total RNAs were extracted from samples of frontal cortex, spinal cord or skeletal muscle by using TRI Reagent LS method (Sigma-Aldrich). RNA (1.5 µg) was used to synthesise cDNA by using random hexamers following the manufacturer’s instructions (SuperScript III reverse Transcriptase; Invitrogen) in a final volume of 20 µL. PCR amplification was carried out using primers chosen in exons of SCN1A (5′-GAAGAACAGCCCGTAGTGGAA-3′; 5′-TTCAAATGCCAGAGCACCA-3′) as previously described.12 RT-PCR products were separated by 2% agarose gel electrophoresis and labelled with ethidium bromide. As internal control for PCR amplification, β-actin cDNA was co-amplified (5′-GCCAGGTCATCACCATTGG-3′; 5′-AGGACTCCATGCCCAGGAA-3′).
Results
Identification of de novo heterozygous missense SCN1A variants in arthrogryposis
In family 1, the affected child was born to non-consanguineous healthy parents (figure 1). During pregnancy and based on ultrasound examination, the male fetus displayed from 23 weeks of gestation (w.g.) bilateral flexion of both hands, a hyperextension of knees and reduced swallowing. These data were confirmed at the third trimester ultrasound examination. The child was born at 38 w.g. and displayed microretrognathism, distal joint contractures of both lower and upper limbs, severe hypotonia and respiratory distress (figure 2). From postnatal day 2, he developed drug-resistant epileptic seizures. Six video EEGs were performed between the age of 2 and 15 days of life, showing dysmature aspect with high amplitude temporal and central delta brushes and discontinuous (up to 15 s of inter-burst interval) and intermittent asynchronous activity in quiet sleep without typical aspect of suppression burst. Frequent rolandic and temporal sharp waves and bi-occipital and central seizures were recorded with subtle clinical features (figure 3). Brain MRI, metabolic investigation and array CGH were normal. Electromyography (EMG) and nerve conduction velocity studies revealed no evidence for peripheral neuropathy nor motor neuron involvement. Morphological analysis of muscle biopsy did not reveal any sign suggesting a congenital myopathy or muscle denervation. The patient was sequentially treated by the following drugs without efficacy on seizure control: phenobarbital, phenytoin, pyridoxine and clonazepam. The newborn died at 21 days from complete ventilator dependency and intractable epilepsy. WES was performed from DNA of the index case. A heterozygous variant was identified in SCN1A in the affected individual of family 1 (figure 1). It is a missense variant (NM_001165963.2:c.2679A>C) predicted to be pathogenic with a high score (p.Leu893Phe; Polyphen-2 score of 1). This variant is not annotated in dbSNP153, 1000 Genomes, ExAC nor TOPMED databases. The variant was confirmed by Sanger sequencing using primers flanking the variant in the affected child. Analysis of both parents showed that the variant had occurred de novo (figure 1).

De novo variants in SCN1A in three arthrogryposis multiplex congenita families and transcript analysis. (A) Pedigrees with Sanger sequencing results for families 1, 2 and 3 are shown. Arrows indicate mutant nucleotide positions. The affected individuals carry de novo heterozygous SCN1A variants. The nucleotide and amino acid changes based on NM_001165963.2 and NP_001159435.1 reference sequences respectively are indicated. Open symbols: unaffected; filled symbols: affected. (B) RT-PCR amplification was performed using SCN1A specific primers on RNAs extracted from the brain motor cortex (BR), spinal cord (SC) and skeletal muscle (SM) of human fetuses at 27 or 22 w.g. When compared with the β-actin control, SCN1A is expressed in brain and spinal cord but not in skeletal muscle. MW, molecular weight.
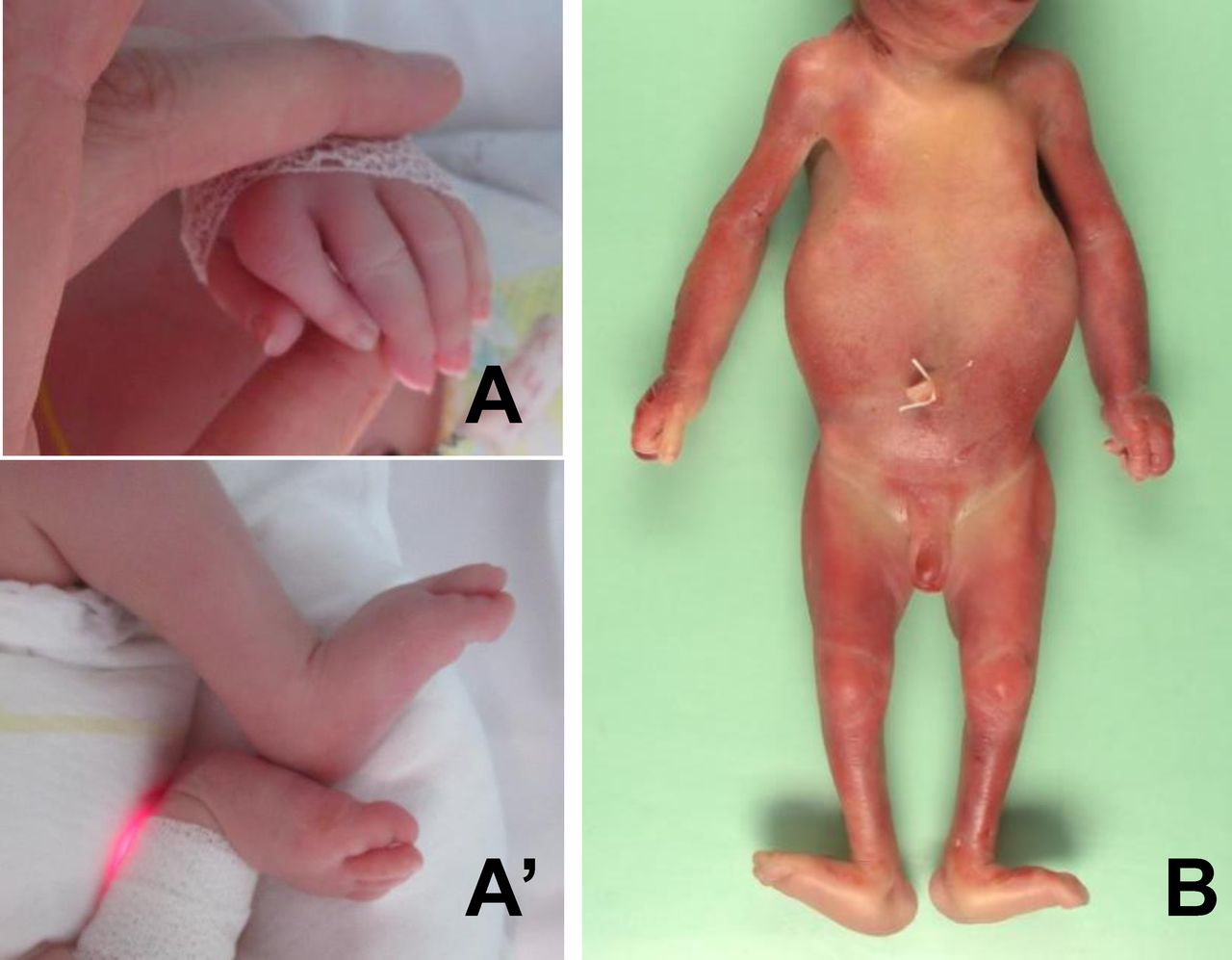
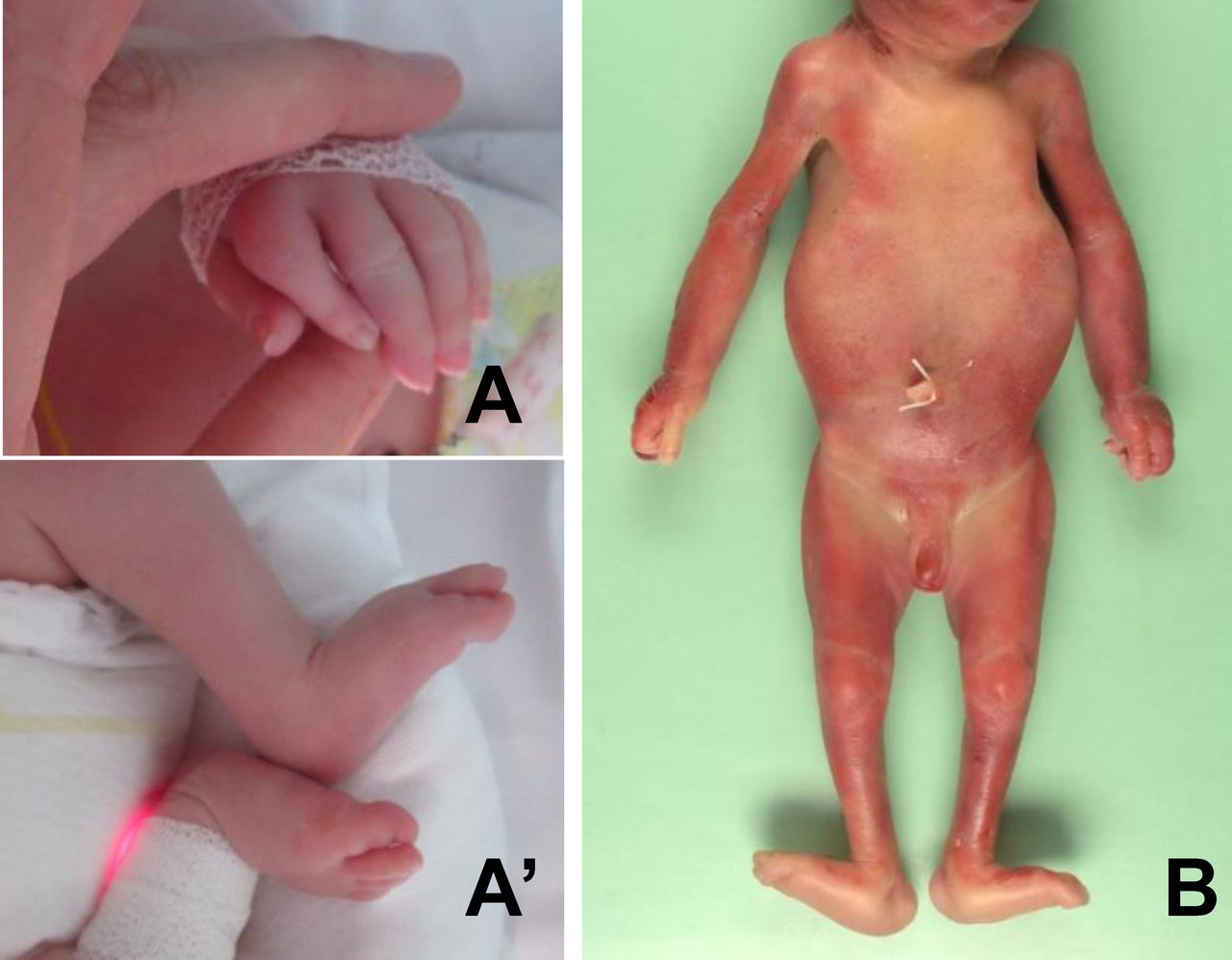
Arthrogryposis multiplex congenita phenotype of the affected individuals. (A, A′) The affected child of family 1 displayed distal joint contractures including camptodactyly and dorsal flexion of feet. (B) Postmortem examination of the affected fetus of family 3 confirmed bilateral camptodactyly, hyperextension of knees and hallux valgus of feet.

Inter-ictal and ictal EEG at day 2 of patient 1. (A) Active sleep: frontal sharp transients and continuous slow-wave activity mixed with abundant rapid rhythms predominating on central regions (Delta brushes). (B) Quiet sleep with discontinuous activity and bursts composed of high-amplitude slow waves mixed with rapid rhythms and sharp waves. (C) Ictal EEG with seizures beginning on right occipital region, spreading to left occipital and bi-central regions.
Analysis of WES previously performed on the DNA samples from additional affected individuals with AMC and/or reduced fetal mobility of unknown origin (n=147) was revised and allowed the identification of predicted pathogenic heterozygous variants in SCN1A in two additional individuals. In family 2, based on ultrasound examination at 25 w.g., the male fetus displayed a lack of fetal movements associated with arthrogryposis of the upper limbs and microretrognathism. The pregnancy was terminated at 25 w.g. at the request of the parents. Postmortem examination confirmed micrognathia without cleft palate, generalised oedema, very short neck, hyperextension of both legs in front of the trunk, dorsal flexion of feet, bilateral camptodactyly with flexion of elbows, right diaphragmatic hernia and marked dorsolumbar scoliosis. Neuropathological examination did not reveal any brain or spinal cord abnormalities. In the affected fetus of family 2, a heterozygous missense variant (NM_001165963.2:c.2965G>A) predicted to be pathogenic with a high score (p.Ala989Thr; Polyphen-2 score of 1) was identified. This variant is not annotated in databases and was confirmed by Sanger sequencing using primers flanking the variant in the affected fetus. Analysis of both parents revealed that the variant had occurred de novo (figure 1).
In family 3 (figure 1), based on ultrasound examination performed at 21 w.g., the male fetus displayed bilateral camptodactyly, hyperextension of knees and hallux valgus of feet. The pregnancy was terminated at 24 w.g. at the request of the parents. Postmortem examination confirmed the ultrasound findings (figure 2). Neuropathological examination did not reveal brain nor spinal cord abnormalities. A heterozygous missense variant (NM_001165963.2:c.707T>C) predicted to be pathogenic with a high score (p.Ile236Thr; Polyphen-2 score of 0.99) was identified in the affected fetus. This variant is annotated in dbSNP153 (rs886039464) database with unknown frequency. This variant was confirmed by Sanger sequencing using primers flanking the variant in the affected fetus. Analysis of both parents showed that the variant had occurred de novo (figure 1).
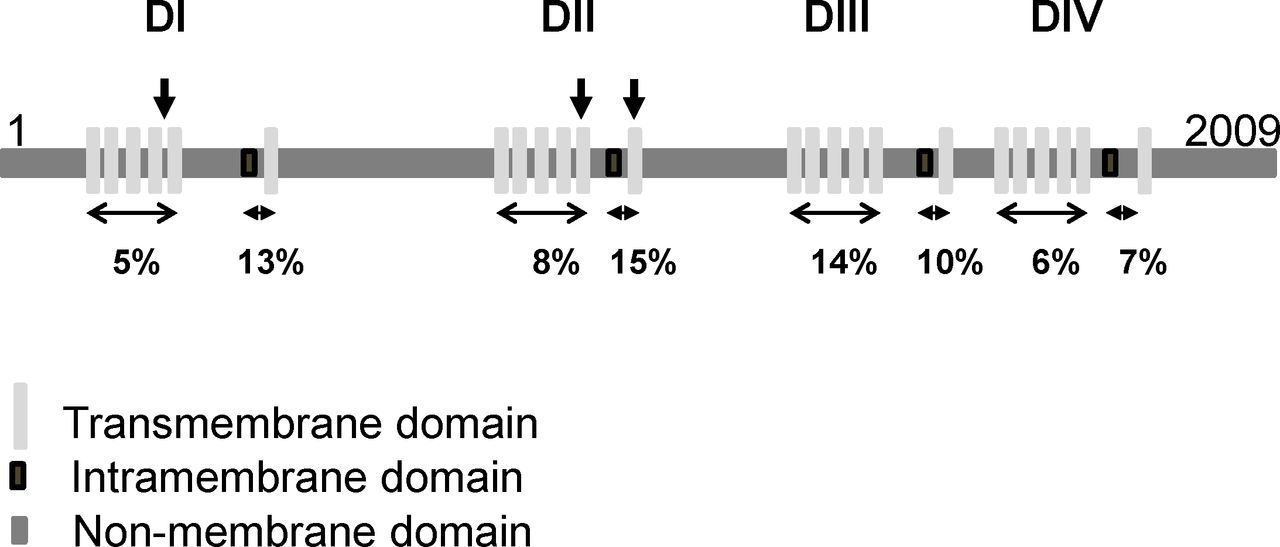
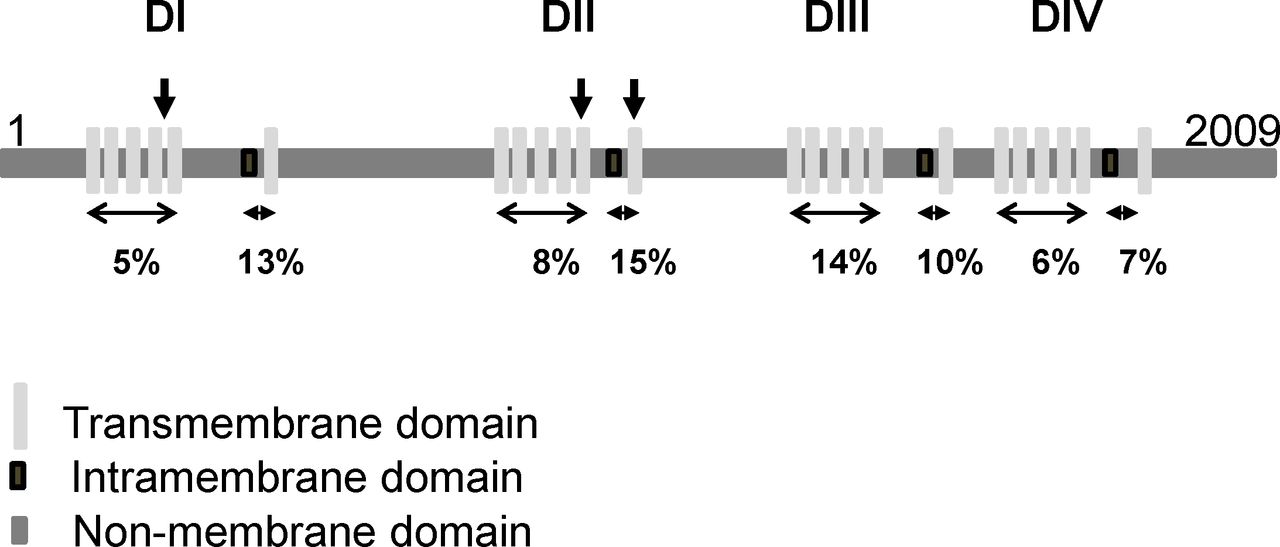
Looking at the position of variants of SCN1A in the three affected individuals did not reveal a specific domain of the Nav1.1 protein when compared with reported de novo pathogenic variants of SCN1A in Dravet syndrome (figure 4, Clinvar, NCBI). Since the disease-causing mechanism is a loss-of-function in a single copy of the SCN1A gene in Dravet syndrome, allelic variants of SCN1A could be responsible for more severe phenotypes. Through re-analysis of WES data and real-time PCR performed on DNA of affected individuals using primers located in exons 1, 10, 14, 15 and 16 of SCN1A, no additional variant including insertion/deletion in SCN1A was detected in the three affected individuals.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Protein structure of SCN1A and distribution of disease-causing missense variants found in patients with arthrogryposis multiplex congenita (AMC) compared with those found in Dravet syndrome. SCN1A is a monomer and consists of four homologous domains (DI–DIV). Each domain has six transmembrane and an intramembrane domains (the central pore). The positions of variants that cause AMC are indicated by vertical arrows. The percentage indicates the proportion of reported de novo pathogenic variants relative to the size of each domain and found in Dravet syndrome.
Mice with heterozygous deletion of Scn1a (Scn1a+/−) model a number of Dravet syndrome features, including spontaneous seizures and premature lethality. Phenotypic severity in Scn1a+/− mice strongly depends on strain background.13 On the 129S6/SvEvTac strain, Scn1a+/− mice exhibit no overt phenotype, while on the (C57BL/6J×129S6/SvEvTac)F1 strain, Scn1a+/− mice exhibit spontaneous seizures and early lethality suggesting modifier loci.13 Using genetic mapping on backcrosses, Miller et al 13 identified several candidate loci that influence premature lethality of Scn1a+/− mice. Differential expression of genes within these candidate loci have been reported including RELN, LGI2, GABRA2, MAPK10, ATP1A3, SLC7A10, GABRG3, KCNJ11, GABRB3, CLCN3, CACNA1A, GABRA6 and GABRB2.13 These genes might be regarded as modifiers in the affected AMC individuals reported here. However, we did not identify any predicted pathogenic variant in these candidate genes.
We also looked at genes encoding other components of axon initial segment (AIS)14 under the hypothesis of a loss of compensatory mechanism of genes encoding other components of AIS such as Nav1.2, Nav1.6 to 1.9, or NALCN or CACNA1E, two genes encoding cation channels and reported as responsible for AMC resulting from central nervous involvement.4 5 No predicted pathogenic variants in these genes were identified in the affected AMC individuals. Finally, we also tested the hypothesis of modifying genes unrelated to these genes. To this end, reanalysis of WES of the three affected individuals was performed. No predicted pathogenic variant in genes shared by the affected individuals except for SCN1A was identified. A larger number of affected individuals is likely required to isolate relevant modifier signal.
Arthrogryposis caused by SCN1A mutation results from brain involvement
SCN1A encodes Nav1.1 belonging to voltage-dependent sodium channels that regulate sodium exchange in excitable tissues including brain, spinal cord and muscle.15 Duflocq et al 16 showed that Nav1.1 expression is concentrated in the proximal AIS and nodes of Ranvier throughout the mouse spinal cord and in many brain regions including cerebral cortex, basal ganglia and cerebellum. The AIS is a specialised structure in neurons that resides between axonal and somatodendritic domains. It serves two functions: (1) to integrate synaptic inputs and generate action potentials and (2) to maintain neuron polarity.17 The myelin sheath produced by Schwann cells and oligodendrocytes attaches to the axon at both sides of the Nodes of Ranvier creating specialised and regularly spaced domains. The propagation of action potentials in myelinated axons requires the precise localisation of Na+ channels at the nodes of Ranvier. These specialised domains underlie saltatory conduction of action potentials along myelinated axons, an essential process for neuronal function.14 To determine whether the AMC phenotype resulted from brain, spinal cord or skeletal muscle defect, SCN1A RNA expression analysis was performed from brain motor cortex, spinal cord or skeletal muscle samples from 22 w.g. and 27 w.g. human fetuses affected from unrelated lethal disorders. RT-PCR analysis revealed an expression of SCN1A in brain and spinal cord but not skeletal muscle when compared with β-actin expression at w.g. similar to the age of AMC detection in the affected fetuses (figure 1). The RNA expression of SCN1A was similar to that reported in GTEx Portal (GTEx Analysis Release V8).
Gitiaux et al 18 reported several cases with Dravet syndrome and EMG recordings showed features of chronic denervation. However, in case 1, EMG and nerve conduction velocity did not reveal any reduction of nerve conduction velocity nor features consistent with chronic denervation. Consistently, Spagnoli et al 19 reported a case with severe, pharmacoresistant epileptic encephalopathy with multiple seizure types including tonic spasms associated with severe motor defect including contractures and talipes. Brain MRI and EMG were also normal.
These data strongly suggest that the AMC reported in these three individuals is likely the consequence of brain involvement. In addition, the normal nerve conduction velocity performed in case 1 indicates the absence of node of Ranvier defect of the peripheral nerve in contrast to AMC linked to variants in either GLDN (gliomedin, [MIM: 608603]) or CNTNAP1 (contactin-associated protein 1, [MIM: 602346]) encoding essential components of the nodes of Ranvier and paranodes, respectively, and showing marked defect of peripheral nerve.20 21
The expression pattern of SCN1A as well as the phenotype of the affected fetuses indicate that SCN1A has a critical role during prenatal motor development likely linked to the role of Nav1.1 in motor cortex.
Discussion
Variants in Nav1.1 cause several related disorders including (1) Dravet syndrome, an early-onset epileptic encephalopathy (EIEE6, [MIM: 607208])6, (2) generalised epilepsy with febrile seizures plus (GEFSP2, [MIM: 604403])22 and (3) familial hemiplegic migraine as the main phenotypes (FHM3, [MIM: 609634]).23 In Dravet syndrome which represents the most severe disease phenotype, patients often present with drug-resistant epilepsy beginning in the first year of life. In addition, patients with Dravet syndrome may also suffer from various symptoms including intellectual disability, and behavioural and motor impairment. The incidence of Dravet syndrome is 1 of 15 500.24 Surprisingly, extensive neuropathological examination has not shown any consistent cerebral or spinal cord structural abnormalities, cell loss or neurodegenerative process in this condition.25
Our approach allowed the identification of de novo missense SCN1A variants in three unrelated individuals with AMC. This conclusion is strongly supported by multiple lines of evidence: the variants are de novo and are absent from various control databases; the variants occur at residues that exhibit a high level of evolutionary conservation. In one patient, the phenotype was characterised by the association of AMC with severe and early-onset drug-resistant epileptic seizures and abnormal EEG background activity. The abnormal motor phenotype was detected from the second trimester of pregnancy and characterised by AMC in the three affected individuals. SCN1A expression during human development, EMG and morphological analysis of the affected individuals strongly suggest that AMC is linked to SCN1A defect in motor cortex.
The expression pattern of SCN1A in the central nervous system during development and the fetal onset of motor defect associated with missense variants of SCN1A reported here strongly suggest that SCN1A has an essential role in prenatal motor development. Our data show that variants of SCN1A encoding essential components of the AIS are responsible for severe developmental motor defects leading to AMC in addition to epilepsy. Taken together, the results of our study combined with previous investigations suggest a large spectrum of SCN1A-related phenotypes ranging from early-onset epileptic encephalopathy to fetal onset of motor defect leading to arthrogryposis.
SCN1A variants in Dravet syndrome have been mainly associated with loss-of-function properties. A more severe and earlier onset phenotype has been recently described, associated with p.Thr226Met variant and gain of function properties.26 Importantly, in family 1, both arthrogryposis and early-onset epileptic encephalopathy were observed which suggests that the pathogenic mechanism of the identified missense mutation (p.Leu893Phe) may be more deleterious in our patients leading to severe motor defect in addition to early-onset epilepsy. Similarly, in the patient reported by Spagnoli et al,19 both severe motor defect and epilepsy were observed: this patient carries a de novo SCN1A missense mutation (p.Ser228Pro) which reinforces the hypothesis of the same but more severe pathogenic mechanism in this new disease phenotype.
Interestingly, two other genes encoding channels are responsible for similar clinical features. NALCN encoding a G-protein–coupled receptor-activated channel is highly expressed in the central nervous system and de novo variants of NALCN are responsible for AMC associated with developmental delay (CLIFAHDD, [MIM: 616266]).4 Epilepsy was reported in two out of seven affected AMC individuals.4 Similarly, CACNA1E encodes the alpha1-subunit of the voltage-gated Cav2.3 channel which is highly expressed in the central nervous system. De novo variants of this gene are responsible for early-onset pharmaco-resistant seizures (EIEE69, [MIM: 618285]) and 43% of patients had congenital joint contractures.5 Altogether, our data enlarge the group of AMC linked to central nervous system involvement caused by variants of genes encoding channels indicating a critical role of these genes in fetal motor development as reported in other developmental channelopathies responsible for cerebral cortex malformations.27
The identification of SCN1A pathogenic variants should allow critical information of families on the large clinical spectrum in addition to AMC, such as early-onset epileptic encephalopathy, and the need for careful postnatal follow-up of the affected individuals in order to propose optimal care of both AMC and epilepsy.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplementary information. All data relevant to the study are included in the article and are available.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors DJ, DB and MVS conducted molecular analyses. CG, SB, FM, JS, CF-B, JMa and AL recruited and phenotyped the patients. CG conducted electrophysiological studies and contributed to writing the manuscript. AK conducted EEG studies and contributed to writing the manuscript. AL contributed to writing the manuscript. JMe designed the study, performed bioinformatics analysis of whole-exome sequencing data and wrote the manuscript. All authors reviewed the manuscript.
Funding This work was supported by a grant from the Association Française contre les Myopathies (AFM, R16070LL), the Agence de Biomedecine (2016) and the Institut National de la Santé et de la Recherche Médicale (Inserm) to JMe.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.