Article Text

Abstract
Background Monogenic hypertension describe a series of hypertensive syndromes that are inherited by Mendelian laws. Sometimes genetic testing is required to provide evidence for their diagnoses, precise classification and targeted treatment. This study is the first to investigate the clinical utility of a causative gene screening and the combined yield of gene product expression analyses in cases with suspected monogenic hypertension.
Methods We performed a large-scale multi-centre clinical genetic research of 1179 expertly selected hypertensive individuals from the Chinese Han population. Targeted sequencing were performed to evaluate 37 causative genes of potential cases of monogenic hypertension. Pathogenic and likely pathogenic variants were classified using the American College of Medical Genetics guidelines. Additionally, 49 variants of unknown significance (VUS) that had relatively high pathogenicity were selected and analysed using immunoblot protein expression assays.
Results 21 pathogenic or likely pathogenic variants were identified in 33 of 1179 cases (2.80%). Gene product expression analyses showed 27 VUSs harboured by 49 individuals (4.16%) could lead to abnormally expressed protein levels. Consequently, combining genetic screening with gene product expression analyses increased the diagnostic yield from 2.80% to 6.79%. The main aetiologies established were primary aldosteronism (PA; 27, 2.29%) and pheochromocytoma and paraganglioma (PPGL; 10, 0.85%).
Conclusion Molecular diagnoses obtained using causative gene screening combined with gene product expression analyses initially achieved a modest diagnostic yield. Our data highlight the predominant roles of PA and PPGL. Furthermore, we provide evidence indicating the enhanced diagnostic ability of combined genetic and functional evaluation.
- clinical genetics
- diagnosis
- hypertension
- genetic screening/counselling
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Among the causes of secondary hypertension are a group of disorders with a Mendelian inheritance pattern that are recognised as monogenic forms of hypertension.1 Monogenic hypertension frequently results in arterial hypertension, electrolyte and hormonal abnormalities,2 3 and drug-resistance4 and often leads to higher risk of cardiovascular events and premature death.5
Monogenic hypertension has been genetically dissected and a total of 37 pathogenic genes of 14 forms of monogenic hypertension have been identified6 7 (online supplementary table 1). Unlike primary hypertension, many monogenic forms of hypertension are amendable if precise diagnoses are obtained and causal interventions are prescribed.1 Recently published 2018 ESC/ESH Guidelines recommended that genetic testing should be considered in specialist centres for patients suspected to have monogenic causes of secondary hypertension.8 However, large-scale genetic screenings for all currently known monogenic hypertension subtypes in hypertensive population is lacking.
Supplemental material
To obtain representative findings, we conducted a Chinese Han population-based, multi-centre genetic screening to investigate the landscape of monogenic hypertension in both clinical and genetic aspects by using a targeted sequencing gene panel. We took three major steps in the development and application of this diagnostic system. First, 37 pathogenic genes for 14 monogenic forms of hypertension were included in the gene panel. Second, targeted sequencing in 1179 individuals suspected of having monogenic hypertension were performed. Third, 49 of the 570 variants of unknown significance (VUSs) were selected to undergo gene products expression analyses. The combination of genetic testing and expression analyses achieved a diagnostic yield of 6.79%.
Patients and methods
Patients and controls
From June 2016 to February 2017, subjects were recruited from hospitals covering multiple regions of China. Hypertensive patients were evaluated by a prescreening work-up to determine whether they meet one of the inclusion criteria and have monogenic hypertension potential. Patients were included if they had at least one of the following items: (1) early onset of hypertension: age of onset ≤35 years; (2) resistant hypertension: systolic blood pressure (SBP) >140 mm Hg and/or diastolic blood pressure (DBP) >90 mm Hg after using ≥three antihypertensive drugs for at least 1 month. Onset age and antihypertensive drug usage were collected from the patients’ medical history; (3) hypertension with electrolyte abnormalities: Blood potassium: 3.5–5.5 mmol/L; Blood sodium: 135–145 mmol/L; Blood chlorine: 96–106 mmol/L; Plasma PH: 7.35–7.45; (4) hypertension with abnormal hormone levels: the normal ranges of aldosterone, renin,9 catecholamine and its metabolites,10 as well as sex hormones11 are documented in relevant guidelines, (5) hypertension with abnormal imaging results: adrenal or abdominal CT scan was performed to investigate the existence of adrenal tumours or other abdominal tumours, or (6) hypertension with special signs: central obesity, moon-face, cafe au lait macules, abnormal sexual development, etc. Physical examination was performed to identify special signs. Patients were excluded if they had other known causes of secondary hypertension other than monogenic hypertension (online supplementary methods 1.1). Trained clinicians were required to evaluate electronic health records of patients to determine patients’ inclusion and exclusion. An external control population (1256 healthy individuals) was introduced to help in filtering out variants that may have low pathogenicity (online supplementary methods 1.1).
Blood pressure (BP) measurement
BP was measured between 7:00 and 9:00. Individuals were asked to refrain from smoking and drinking tea or coffee for more than 30 min and to sit and rest for 15 min before measurement. Right brachial artery BP was measured. SBP was recorded on hearing the phase I Korotkoff sound. DBP was recorded on hearing the phase V Korotkoff sound. BP was measured two times with a 30 s interval. If two measurements differed by >5 mm Hg, BP was re-measured. Final BP was calculated as the mean of two or three measurements. Hypertension was defined as SBP ≥140 mm Hg and/or DBP ≥90 mm Hg; or BP <140/90 mm Hg with regular antihypertensive drugs usage.
Pathogenic genes selection and panel sequencing
The 37 known pathogenic genes were well documented in OMIM or Human Gene Mutation Database and exhibit causal relationships with monogenic forms of hypertension (See online supplementary table 1 and the references cited). Genome DNA were obtained from oral swab samples. Fragment libraries were constructed following the Agilent standard library preparation protocol for TruSeq (Illumina). The resulting libraries were sequenced on a HiSeq 4000 platform (Illumina, San Diego, California, USA) according to the manufacturer’s instructions. Fast-format reads were aligned to the human reference genome (GRCh37/hg19) using BWA-0.7.10 aligner12 (online supplementary methods 1.2).
Annotation, filtering and classification of variants
Variant annotation was performed using ANNOVAR.13 The allele frequency of variants was annotated in the Exome Aggregation Consortium (ExAC). Pathogenicity was assessed using SIFT (V.6.2.1),14 Polyphen, MutationTaster,15 and CADD.16 Potential disease causing variants were required to meet: (1) located in exonic region or within 2 bp of intron–exon boundaries of the 37 genes, (2) allele frequency <0.01 in the ExAC database, (3) predicted to be deleterious or with no prediction result, (4) CADD >10, (5) detected in ≤1 control sample. We excluded loss function variants identified from genes that gain of function mutations were known mechanisms and gain of function variants identified from genes that loss of function (LoF) mutations were known mechanisms.
Interpretation of variants reported by panel sequencing
American College of Medical Genetics (ACMG) guidelines were used to determine pathogenicity of variants.17 Variants were classified as five categories: (1) pathogenic, (2) likely pathogenic, (3) benign, (4) likely benign and (5) VUSs. Each variant was manually evaluated by three independent interpreters who then agreed by consensus. To facilitate the identification of potential deleterious VUSs, we listed the ACMG evidence terms of them and then referred to the clinical phenotypes of patients. VUSs that have more pathogenic evidence terms (indicating higher pathogenicity) and match their clinical phenotypes were selected as candidate variants for gene product expression analyses.
Gene product expression analyses
293A cells were transfected with recombinant plasmids of Flag-gene (wild type) and Flag-gene (mutant) using Lipofectamine 3000 Reagent (ThermoFisher) according to the manufacturer’s instructions. Identical transfection efficiencies were confirmed by comparing the amount of green fluorescence in cell cultures. mRNA levels were estimated by real time QPCR (ViiA, Applied Biosystems, USA). The expression of target genes was normalised to the mRNA level of GAPDH as an internal control and the mRNA expression of different groups was normalised to 2−ΔΔCt. Protein levels were determined by immunoblotting with anti-Flag antibodies (Abcam, Cambridge, UK). Mouse monoclonal anti-β-actin antibody was used to confirm equal loading of cellular proteins. The immune complexes were visualised and detected using Tanon 5200 chemiluminescent imaging system (Shanghai, China). Greyscale bands were quantified using Image J software (V.d 1.46). The mRNA (2−ΔΔCt) and protein levels (gray-scale) between wide-type and mutant genes were compared by unpaired two-tailed Student’s t test. The significance level was set to p<0.05. See online supplementary methods 1.3 for detailed information.
Statistical analysis
Comparison was performed using the Student t-test for normally distributed continuous variables, the Mann-Whitney U test for non-normally distributed continuous variables, and the χ2 test for categorical variables. All statistical analyses were performed using R (V.3.2.1.) and p<0.05 was used to indicate statistical.
Results
Clinical characteristics of study population
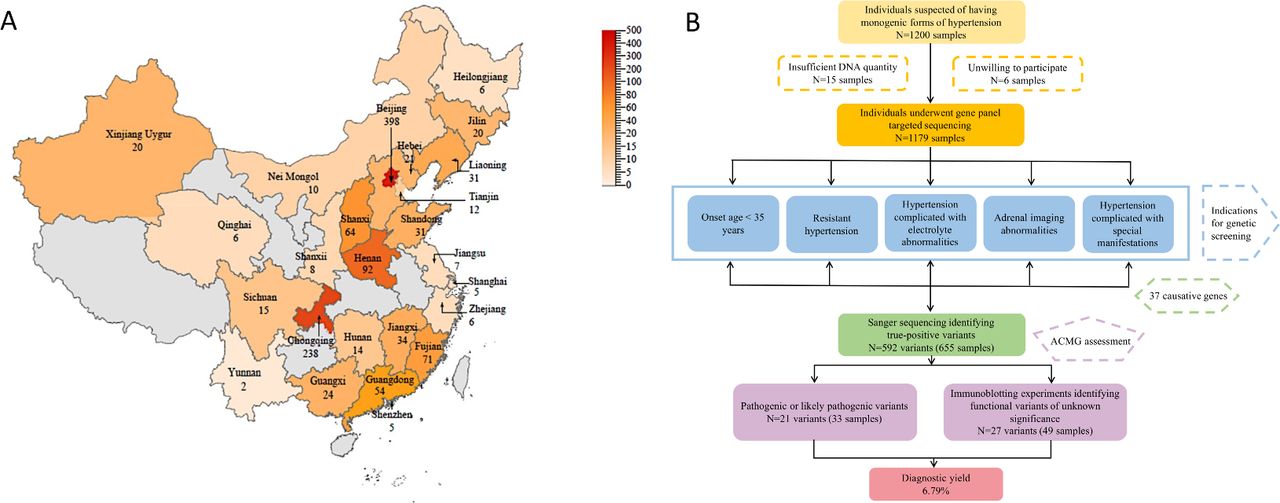
One thousand two hundred hypertensive individuals suspected of having monogenic hypertension were enrolled from 71 hospitals covering 19 provinces, four municipalities and three autonomous regions of China. Twenty-one individuals were excluded for low-quality DNA, insufficient quantities of DNA, or unwillingness to participate in this study (figure 1). Genetic screening was performed in 1179 patients. Of them, 1039 had complete clinical information. The mean age of onset was 35.21 years. Two hundred and twenty-seven (21.85%) were suspected to have primary aldosteronism (PA), 76 (7.31%) were suspected to pheochromocytoma and paraganglioma (PPGL), 18 (1.73%) were suspected to have Cushing syndrome, 4 (0.38%) were suspected to have Liddle syndrome, while the rest individuals had no suspected diagnosis (online supplementary table 2).

Sample distribution and flow chart of the current study. (A) Sample distribution in provinces of China. The number of samples in each province is represented by different shades of orange. Dark shade represents more samples, while light shade represents less samples. (B) A flow chart of the current study. ACMG, American College of Medical Genetics.
The mutational landscape of individuals
After variants filtering, 598 variants harboured by 661 individuals were identified. Sanger sequencing was then performed to exclude false-positive variants. Five hundred and ninety-two variants harboured by 655 (55.56%) individuals were retained, whereas the remaining 524 patients (44.44%) had no variants (table 1). The most frequently mutated gene was CACNA1H (online supplementary figure 1). Sixty-two of the 592 variants were shared by ≥3 individuals (online supplementary table 3). Five of the 62 were identified from pedigrees and resulted in shared phenotypes. The remaining 57 variants were identified from unrelated individuals and two of them could lead to similar phenotypes (online supplementary table 4). No consanguineous marriage was observed.
Summary of the results of gene panel targeted sequencing in 1179 individuals
According to ACMG guidelines, 21 variants were found to be pathogenic or likely pathogenic (table 2), which may be responsible for monogenic forms of hypertension. Nine of the 21 were null variants that can be scored as pathogenic very strong and may lead to absence of gene products. The 21 variants were harboured by 33 individuals, and thus, the diagnostic yield of the current stage was 2.80%. None of the 33 individuals carried more than one pathogenic or likely pathogenic variants and all of these variants were absent from the control dataset. Five hundred and seventy variants were considered to be VUSs, whose clinical significance remained unknown. Of note, we identified 18 stop-gain variants and 13 frame-shift variants (online supplementary table 5). Among them, three stop-gain (ARMC5, CYP17A1 and SDHD) and three frame-shift variants (MEN1, SCNN1B and VHL) were classified as pathogenic or likely pathogenic. LoF variants in all of the six genes are known mechanism of hypertension.18–23 Additionally, in four of the six genes (ARMC5, 24 SDHD,25 MEN1 26 and VHL 27), stop-gain or frame-shift variants were proved to be targets of nonsense-mediated mRNA decay.
Overview of the 21 pathogenic or likely pathogenic variants identified by gene panel targeted sequencing
Phenotypes and genetic findings of individuals with multiple locus variants
In 655 individuals with positive findings, 131 (20%) harboured mutations in ≥2 genetic loci. We found three patients could be diagnosed with two disorders for harbouring variants from two distinct loci (online supplementary table 6). The first patient was a compound heterozygote of two CYP17A1 variants with 17-alpha-hydroxylase deficiency manifestations. She also carried a predicted deleterious heterozygous SCNN1B variant. After glucocorticoids and sex hormones treatment, her hypertension and hypokalemia were partly controlled. Amiloli experimental treatment was administered and her hypertension and hypokalemia were ideally controlled. The second patient carried a heterozygous variant of SCNN1G and a heterozygous variant of ARMC5 with Cushing syndrome manifestations. Unilateral adrenalectomy partly controlled his BP and cortisol level. Amiloli was further administered and his hypertension and hypokalemia were completely controlled. The third patient carried a heterozygous variant of CACNA1H and a heterozygous variant of WNK4 with pseudohypoaldosteronism, type IIB manifestations except for normal blood potassium. However, he had left adrenal nodules and aldosterone to renin ratio (ARR) >30, indicating the existence of PA. The patients’ normal blood potassium may be due to the fact that pseudohypoaldosteronism, type IIB could lead to hyperkalemia, while PA could result in hypokalemia.
Gene product expression analyses of VUSs
Forty-seven VUSs in 17 causative genes were selected for gene product expression analyses (online supplementary figure 2). Although some variants were predicted to be LoF variants, expression analyses were still performed to confirm their roles in 293A cell lines. Equal transfection efficiencies between wild type and mutant groups were confirmed (online supplementary figure 2) and the mRNA levels between wild type and mutant genes showed no statistical difference in 16 genes. However, two stop gain variants in ARMC5 and VHL resulted in significantly reduced mRNA levels (online supplementary figure 3), which may be due to the nonsense-mediated mRNA decay. Nineteen of the 47 VUSs resulted in reduced protein levels, while eight VUSs resulted in increased protein levels (figure 2, online supplementary figure 4). Therefore, 27 variants were regarded as potential functional VUSs (online supplementary table 8). The 27 potential functional VUSs were either located adjacent to mutation hotspots or located in well documented disease causing sites (online supplementary table 9) and the clinical phenotypes of the 49 patients were inconsistent with their genetic findings (online supplementary table 10). Thus, the general diagnostic rate obtained in the current study rose from 2.80% to 6.79%. None of the 49 individuals carried more than one potential functional variant. However, seven of them carried one potential functional variant and at least one other VUS. Their phenotypes were all consistent with the clinical manifestations of the potential functional variants but not related with VUSs they harboured (online supplementary table 11).

Schematics of mutant proteins and immunoblot analysis for variants of unknown significance. (A) Schematics of mutant proteins. Rare variants identified by panel testing were labelled in schematics. Variants that underwent immunoblot analysis were marked by “#” and functional variants confirmed by immunoblot analyses were highlighted by red font. (B) Immunoblot analysis for variants of unknown significance. All of the immunoblot analysis results for 49 variants that underwent gene product expression analyses were shown in online supplementary figure 4.
Associations between clinical characteristics and genetic testing results
We totally identified 33 individuals with pathogenic or likely pathogenic variants, 49 individuals with potential functional variants, 572 individuals with VUSs, and one person with a benign variant (figure 3A, table 2). Diseases that harboured the greatest burden of pathogenic, likely pathogenic or potential functional variants were PA and PPGL (figure 3B). The BP of individuals with pathogenic and likely pathogenic variants were clustered predominantly in the 150–170/95–105 mm Hg interval. Interestingly, individuals with potential functional variants were also distributed in the same interval, while individuals in the remaining three groups were distributed differently (figure 4A). The age of onset of individuals with pathogenic and likely pathogenic variants were the youngest among all groups and mainly clustered in the 1–15 years’ interval. Moreover, patients with potential functional variants were slightly younger than individuals in the rest three groups (figure 4B).

Summary of the results of gene panel targeted sequencing. (A) Genetic identification of 1179 individuals. (B) Variants distribution in different forms of monogenic hypertension. In 4B, the vertical axis and the column on the left side represent the total samples with positive panel results, whereas the vertical axis and the column on the right side represent samples carrying pathogenic, likely pathogenic and potential functional variants identified by gene product expression analyses.

BP levels and age distributions in individuals who underwent genetic screening. (A) BP levels distribution in individuals who underwent genetic screening. (B) Age distribution in individuals who underwent genetic screening. The vertical axis represents the ratio of the sample number in each BP or onset age group to the total sample number of group with positive panel results (total), with P and LP variants, with PF variants, with VUSs, or with negative and benign variants. B, benign; BP, blood pressure; LP, likely pathogenic; N, negative; P, pathogenic; PF, potential functional; VUS, variants of unknown significance.
Genetic screening of family members
Five pedigrees were observed in the current study (figure 5). Pedigree analyses uncovered a known pathogenic variant responsible for MEN2B (Family 1, RET c.T2753C), two novel pathogenic variants for pre-eclampsia (Families 2 and 3, CACNA1D c.A920G and c.G4370A p.R1457Q), a novel pathogenic variant for Liddle syndrome (Family 4, SCNN1B c.C1513T) and a known pathogenic variant for 17α-hydroxylase deficiency syndrome (Family 5, CYP17A1 c.985_987delinsAA). See online supplementary information for detailed information. CACNA1D variants have been well documented to be a causative gene responsible for PA. However, in family 2 and family 3, the probands and their affected relatives with CACNA1D mutations cannot be diagnosed as PA for failing to surpass the screening test ARR or confirmatory test. This suggest that CACNA1D variants identified in the current study may be responsible for pre-eclampsia rather than PA. These identifications expended the phenotypic profiles related with CACNA1D.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Family trees of five pedigrees in the current study. The black triangles indicate probands in the families. Different phenotypes were indicated by distinct markers as shown in the family trees. (A) Multiple endocrine neoplasia family. (B) The first pre-eclampsia family. (C) The second pre-eclampsia family. (D) Liddle syndrome family. (E) 17α-hydroxylase deficiency syndrome family.
Discussion
We report results obtained from causative gene screening combined with gene product expression analyses of a large cohort of individuals suspected of having monogenic hypertension in a clinical setting. A diagnostic yield of 6.79% was achieved by the current study. Genetic testing is undoubtedly of considerable clinical value for individuals suspected to have monogenic hypertension. Actually, genetic testing is the ‘gold standard’ of some monogenic hypertension, such as Liddle syndrome and glucocorticoid-remediable aldosteronism. Sometimes, even the diagnoses have been achieved, genetic testing is still recommended for disease classification and targeted treatment because the hereditary pattern, complicated diseases, growth characteristics and malignancy of monogenic hypertension are largely determined by gene mutation. Although the diagnoses of some PA patients may be achieved by ARR or confirmatory tests, genetic testing is still warranted. It is not rare to see hypertensive individuals with hypokalemia and adrenal imaging abnormalities have critical ARR values or confirmatory test results. Genetic testing may provide further diagnoses cue.
The diagnostic panel was designed to facilitate the diagnoses of hypertensive patients rather than merely restricted to laboratory findings. There exists a two-way communication system between laboratory and clinical settings. Genetic testing reports were ultimately sent to each patient and their doctors in both paper version and electronic version. Once the patients underwent genetic testing, they were ‘chased down’ by their doctors. The doctors were frequently needed to conduct further clinical phenotyping on patients who had positive testing results. Many patients’ phenotypes, especially those with deleterious variants can be reflected by their genetic findings. The identification of these variants frequently corrected fault diagnoses, optimised preventive processes and offered more precise treatment.
Among the 523 individuals with no variant, 83 (15.87%) were suspected to have specific forms of monogenic hypertension. The clinical manifestations of individuals with suspected diagnoses were in consistent with their suspected forms of monogenic disorder. There might be two reasons for failing to detect potential causative variants. First, the variants filtering criteria were so stringent that some of the potential disease causing variants might be excluded. Second, variants in novo pathogenic genes that not yet been discovered cannot be covered by the panel used in the current study.
Targeted sequencing combined with gene product expression analyses uncovered a number of disease causing variants and potential functional variants that were newly associated with monogenic hypertension. Among 21 pathogenic or likely pathogenic variants, three were novo disease causing variants first reported by this study. Among the 27 potential functional variants, 25 were first reported to be potential disease causing variants. These potential functional variants were identified from 15 genes and variants on eight of the 15 genes were proved to lead to completely abolished protein expression levels. Complete loss of gene products is likely to lead to severe abnormalities to human body. However, for the three genes that do not lead to completely abolished expression of mutant proteins (NOS3, KCNJ5 and SDHC), dose-dependent effects were observed in animal models through in vivo and in vitro experiments. The biological functions of organisms can be influenced by even slight reduction of the expression levels in them.28–30
Multiple variants found in a single affected individual is an important concern of high throughput sequencing studies. The prevalence of multiple molecular diagnoses in a single genome ranges from 3.2% to 7.2%.31–34 Blended phenotypes resulting from multilocus variants can be mistaken for new disorders or newly identified phenotypes of known disorders. Among the 131 individuals harbouring multiple genetic locus, the diagnoses in three of them may be based on variants from two distinct disease locus. The identification of multiple locus variants provided useful information on the patients’ diagnoses and treatments since therapeutic strategy merely targets a single disorder were not sufficient to control the symptoms resulted from multiple genetic disorder.
In this study, PA and PPGL were diseases with the greatest burden of pathogenic, likely pathogenic and potential functional variants. Most of the pathogenic or potential disease causing mutations were found in CACNA1H and CACNA1D genes. This finding is consistent with previous reports that CACNA1H and CACNA1D are more likely to be germline mutation genes and are absent in tumour samples.35 36 Approximately 40% of PPGL cases carry a germline mutation in one of 12 known causative genes.37 Similarly, in this study, 76 individuals were included for suspicion of having PPGL and PPGL variants were identified from 24 of them (31.58%). In a retrospective study based on Caucasian series, the relative frequencies of germline mutations in PPGL causative genes were VHL >SDHB > SDHD>RET > NF1 >TMEM127>MAX > SDHC>SDHA > EPAS1 >EGLN1 38. Our results generally showed a similar pattern, with causative variants being most frequently clustered in RET, VHL, SDHB and NF1, suggesting the necessity for screening these frequently mutated genes in clinical settings.
In this study, some of the individuals were included for having adrenal imaging abnormalities. Although some of them may have asymptomatic ‘incidentalomas’, causative gene sequencing is warranted. As documented by epidemiological studies, during the follow-up of non-functional adrenal mass, the size of 25% of incidentaloma will increase and 20% of them could result in over-secretion of hormones, especially those with diameters>3 cm.39 40 Since PA and PPGL are relative common causes of incidentaloma, genetic testing may be useful to offer aetiological cue and help clinicians to make decision about whether to intervention, to closely follow-up, or just to ignore.
Although this study was designed to evaluate the presence of monogenic hypertension in probands, their affected family members also benefited from genetic screening. Since the codon 918 mutations in family one has been implicated to have high penetrance of pheochromocytoma41 and medullary thyroid carcinoma,42 43 children who harbour this mutation have been recommended to have more aggressive treatment regimens.44 Recently, prophylactic surgery of the three children has been reconsidered. Moreover, pedigree analyses also enable novel pathogenic variants and phenotypes to be identified. In our study, there were two pre-eclampsia probands which had several family members who harbour CACNA1D variants and presented with either pre-eclampsia or early onset hypertension. CACNA1D variants have been well documented to cause PA45; however, no evidence implicates the direct correlation between CACNA1D and pre-eclampsia. A recent study documented that the relative mRNA expression of CACNA1D was significantly higher in placental vessels than those in other vessels. Placental vessels were characterised by much weaker responses to MgSO4-mediated vasodilatation compared with non-placental vessels.46 Thus, our study reports for the first time that CACNA1D mutations may cause pre-eclampsia.
Conclusions
Comprehensive genetic screening was performed among potential cases of monogenic hypertension and resulted in a diagnostic yield of 6.79%. Combining genetic analyses with functional evaluation enhanced the diagnostic ability and uncovered large numbers of novel functional variants. Our data also highlighted the predominant roles of PA and PPGL in monogenic hypertension.
Acknowledgments
The authors would like to thank the individuals and their families for their participation in this study.
References
Footnotes
Contributors JC, XY, YF and BG were responsible for the study design and conception. PL, QL and HC contributed to the sample collection. MB and YZ dealt with the clinical data collection and assembly. MB, YZ, SL, LJ, WW, ZC and JZ contributed to the sequencing data analysis and interpretation. MB drafted the manuscript. All authors participated in revising this manuscript. MB submitted the manuscript.
Funding This work was supported by National Natural Science Foundation of China (8167020374) and Beijing Natural Science Foundation (17G10314).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the Institutional Review Board of Peking Union Medical College Fuwai Hospital (ID: 2016GEC012).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.