Article Text

Abstract
Background Primary hereditary microcephaly (MCPH) comprises a large group of autosomal recessive disorders mainly affecting cortical development and resulting in a congenital impairment of brain growth. Despite the identification of >25 causal genes so far, it remains a challenge to distinguish between different MCPH forms at the clinical level.
Methods 7 patients with newly identified mutations in CDK5RAP2 (MCPH3) were investigated by performing prospective, extensive and systematic clinical, MRI, psychomotor, neurosensory and cognitive examinations under similar conditions.
Results All patients displayed neurosensory defects in addition to microcephaly. Small cochlea with incomplete partition type II was found in all cases and was associated with progressive deafness in 4 of them. Furthermore, the CDK5RAP2 protein was specifically identified in the developing cochlea from human fetal tissues. Microphthalmia was also present in all patients along with retinal pigmentation changes and lipofuscin deposits. Finally, hypothalamic anomalies consisting of interhypothalamic adhesions, a congenital midline defect usually associated with holoprosencephaly, was detected in 5 cases.
Conclusion This is the first report indicating that CDK5RAP2 not only governs brain size but also plays a role in ocular and cochlear development and is necessary for hypothalamic nuclear separation at the midline. Our data indicate that CDK5RAP2 should be considered as a potential gene associated with deafness and forme fruste of holoprosencephaly. These children should be given neurosensory follow-up to prevent additional comorbidities and allow them reaching their full educational potential.
Trial registration number NCT01565005.
- CDK5RAP2
- MCPH
- primary microcephaly
- intellectual disability
- retinal alteration
- sensorineural hearing loss
Statistics from Altmetric.com
Introduction
Primary microcephaly (PM) is a rare neurodevelopmental disorder characterised by congenital microcephaly, intellectual disability of variable severity (ID) and occasional epilepsy that may reflect cortical mantle defects.1–3
Clinically, PM includes isolated PM (MicroCephaly Primary Hereditary (MCPH), PM with short stature (Seckel syndrome, microcephalic primordial dwarfism) and syndromic PM (associated with non-neurodevelopmental manifestations). MCPH and Seckel syndrome may be further subdivided by the presence of cortical malformations and/or chorioretinopathy. Although this classification helps managing the differential diagnosis of the patients, it does not reflect underlying mechanisms. Most PMs show autosomal recessive inheritance,1 3 although dominant forms have occasionally been described4–6 and a mitochondrial origin has been proposed.7 To date, >100 genes have been implicated in PM.8
The cyclin-dependent kinase 5 (CDK5) regulatory subunit associated protein-encoding gene (CDK5RAP2; OMIM 608201) is among the first genes initially identified in PM (MCPH3, OMIM 604804).9 CDK5RAP2 regulates CDK5 activity required for centrosome cohesion,10 centriole duplication and attachment of the centrosome to the spindle pole,11 and plays a role in microtubule plus-end dynamics.12 In addition, it is essential for DNA damage response.13 In the developing mouse neocortex, Cdk5rap2 is crucial for the maintenance of apical neural progenitors14 15 as its deficiency leads to spindle orientation defects, premature cell cycle exit and increased cell death during neurogenesis, resulting in a reduction in the final neuron number.15 During human neurogenesis, CDK5RAP2 is widely expressed in the ventricular and subventricular zones at the weeks of gestation WG12,WG18 and in the superficial layers of the neocortex until term.16
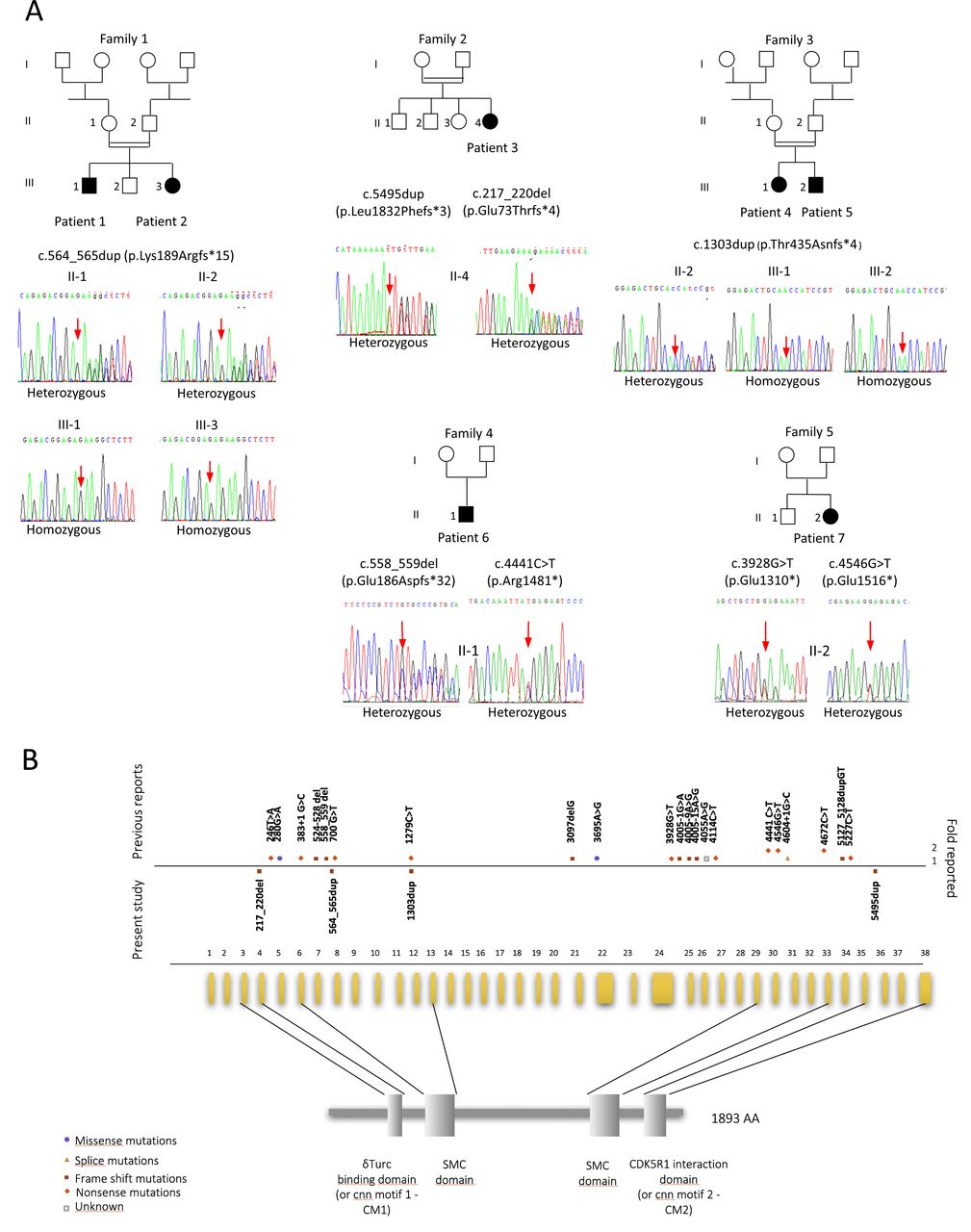
Variants in CDK5RAP2 were first identified in association with PM in 2005.9 Since then, 35 patients from 17 families have been described, essentially through single case or mutation reports, and with limited clinical details.9 17–29 Most mutations reported are nonsense or frameshift variants (ie, null variants), except for three siblings carrying missense variants22 (figure 1 and online supplementary table 1). Patients with pathogenic variants in CDK5RAP2 display heterogeneous phenotypes: the first 11 patients reported are microcephalic individuals (Occipitofrontal circumference OFC from −4 to −9 SD during childhood) with normal stature and mild-to-moderate ID.9 18 26 Three infants were reported with PM and mildly short stature (from −2.5 to −4 SD) that corrected with age. Three additional patients from two pedigrees were reported with Seckel-like microcephalic dwarfism.29 Feeding difficulties,9 20 24 26 skin areas of hypopigmentation9 and/or hyperpigmentation,21 29 seizures9 and deafness9 21 26 29 have been reported. CDK5RAP2 variants have also been reported in patients with corpus callosum agenesis without microcephaly.22 One patient developed acute lymphoblastic leukaemia.9 This heterogeneity is not supported by any genotype/phenotype correlation and the possibility of additional recessive or de novo mutation has not been ruled out.
Supplemental material
Despite this range of presentations, MRI data are lacking. Brain MRI performed in CDK5RAP2 patients have failed to provide specific information. When available, data only indicate a reduced brain size and a qualitative impression of gyral simplification,23 26 28 29 similar to reports in other MCPH.30 Hypoplasia, agenesis or dysplasia of the corpus callosum have been occasionally reported.21 22 24 As MRI data have not been reported for the majority of patients, the possibility that CDK5RAP2 patients have specific brain or cortical malformations cannot be ruled out.
Furthermore, no systematic neuropsychological assessment of the patients has been published. Mild-to-moderate ID was reported in the first cases.9 20 31 However, this finding has been challenged by more recent studies using neuropsychological assessments26 28 surprisingly showing that individuals with CDK5RAP2 variants have a normal/borderline IQ. Hence, the severe reduction in brain volume in patients with CDK5RAP2 variants does not appear to result in an equally severe impact on cognitive functions. There is thus an urgent need for reliable data on cognitive prognosis in MCPH as a prerequisite for genetic counselling, adequate educational therapy and medical follow-up.
Here, we took advantage of a cohort of seven new CDK5RAP2 patients, the largest series reported so far, to revisit the phenotype associated with CDK5RAP2 by performing prospective, extensive and systematic clinical, MRI, psychomotor, neurosensory and cognitive examinations under similar conditions. We report four novel and four recently described CDK5RAP2 mutations7 and show that, in addition to PM, all patients have skin pigmentation changes as well as congenital neurosensory defects, including microphthalmia and retinal pigmentation defects, and a unique cochlear malformation. In agreement with this latter finding, immuno-labelling on human fetal tissues identifies the CDK5RAP2 protein in the developing cochlea, in addition to the fetal brain. Moreover, five patients exhibit a hypothalamic anomaly. Our data indicate that the CDK5RAP2-related phenotype is wider but more homogeneous than previously thought, and that ophthalmic and auditory investigations should be systematically included in the clinical follow-up of these children.
Materials and methods
Next-generation sequencing or targeted sequencing
Homozygosity mapping (patients 1 and 2) was performed by SNP array analysis (SurePrint G3 Human Genome CGH+SNP Microarray kit, Agilent Technologies). Coding exons and splice junctions of CDK5RAP2 were sequenced by standard bidirectional Sanger methods. Mutations in patients 3–5 were identified by targeted next-generation sequencing (NGS). CDK5RAP2 was included on a microcephaly gene panel for cohort screening by multiplex PCR enrichment on a microfluidic support (Access Array, Fluidigm) and 2×150 bp sequencing with an Illumina MiSeq system. Sequencing reads were mapped to the UCSC Genome Browser (hg19) with MiSeq software, the Burrows-Wheeler Aligner and the Genome Analysis Toolkit, and panel exons were 99% covered. Variants were filtered against Mutation Annotation Form (MAF) >0.005, dbSNP, 1000 Genomes, the NHLBI ESP Exome Variant Server and an in-house dataset. Rare variants were annotated for functional features of coding nucleotides with publicly available databases, Polyphen2, SIFT, Mutation taster and Align GVGD. Confirmation of mutations identified by NGS and their familial segregation were carried out (Sanger sequencing). Patients 6 and 7 were part of a whole-exome sequencing study in a larger microcephaly cohort. Methods, sequencing results and few clinical hallmarks of these two patients were reported elsewhere.7
Clinical, neuroimaging and neurosensory assessment of the patients
Seven patients carrying CDK5RAP2 variants identified in Paris or Zurich genetic departments were recruited between 2014 and 2018. Inclusion criteria were: OFC≤−2 SD at birth (WHO growth charts (www.who.int/childgrowth/standards)), no environmental cause of PM. Parents provided informed consent for children’s participation.
The protocol included a detailed clinical and neurological examination and the following exams/assessments:
Cerebral MRI
Included coronal T2-weighted and T1-weighted three-dimensional (3D) sequences with millimetre slices (1.5 T Philips scanner).
Cognitive assessment
Intellectual abilities were assessed using the international Wechsler scales, according to age (Wechsler Intelligence Scale for Children (WISC) IV: 6–16 years, n=5, patients 1, 4–7; Wechsler Preschool and Primary Scale of Intelligence (WPPSI) III: 3–7 years 7 months, n=2, patients 2 and 3). Full-Scale IQ (FSIQ) was calculated from four scores for WISC IV: Verbal Comprehension Index (VCI), Perceptual Organisation Index (POI), Working Memory Index (WMI) and Processing Speed Index (PSI). The scores are independent and involve specific cognitive abilities such as lexical stock, general knowledge, verbal comprehension and verbal reasoning (VCI); visuomotor and visuospatial skills to examine a problem, organise thoughts and find solutions using cubes or pictures (POI); short-term memory, concentration abilities, mental manipulation, planning abilities, cognitive flexibility, arithmetic reasoning (WMI) performance speed in graphic realisation and visual discrimination (PSI). FISQ was calculated from three scores for WPPSI III (verbal IQ, performance IQ, and processing speed quotient). Only the non-verbal reasoning scale has been calculated for patient 3 due to language barrier.
Neurosensory assessment
Ophthalmological assessment included visual acuity measurement with Snellen optotypes axial length measurement, biomicroscopy of the anterior segment and ocular fundus examination. Fundus autofluorescence imaging, full-field scotopic and photopic electroretinograms (ERGs), a pattern-ERG (pERG) and electro-oculography (EOG) were performed in all patients. Two patients underwent optical coherence tomography (3D-OCT).
Audiological assessment included pure tone and speech audiometry.
Temporal bone high-resolution CT included helical acquisition of submillimetre slices with multiplanar reconstructions (Philips Brilliance 64 CT).
Statistical analysis
The four scores required to calculate the FSIQ were compared with search for specific learning disorders (one-way analysis of variance and Bonferroni post hoc test). P values <0.05 were considered to be statistically significant.
Immunohistology in human fetal cochlea and brain
Human fetal temporal bone (1) and brain (2) specimen were obtained from morphologically normal miscarried fetuses (WG12 for (1) and WG8 and WG12 for (2)), with the informed written consent of the parents in accordance with French law. Specimen were fixed and paraffin-embedded and immunohistochemistry was performed using antibody against γTubulin (ab11316, Abcam), Myosin VI (M0691, Sigma) and CDK5RAP2 (A300-554A, Bethyl). Images were acquired using a Confocal Leica SP8.
Results
Molecular findings
All CDK5RAP2 mutations are depicted in figure 1, table 1 and online supplementary table 1.
Clinical and molecular findings of patients with CDK5RAP2 mutations
Neuroradiological findings of patients with CDK5RAP2 mutations

Novel and previously described CDK5RAP2 variants. (A) Pedigree of seven individuals from European (families 2, 4 and 5) or North of Africa’s families (families 1 and 3) and their respective Sanger sequencing chromatograms. (B) Scheme representing novel variants in our series and the known variants in the human CDK5RAP2 gene and corresponding domain of the protein. The variants reported previously are shown above the horizontal line; novel variants from the present study are indicated below this line and just above the exons. Various symbols represent missense, nonsense, splice and frameshift variants as indicated. γTuRC, gamma-tubulin ring complex; CM1, centrosomin (cnn) motif 1; SMC, structural maintenance of chromosomes protein.
Homozygosity mapping performed by SNP array analysis revealed a common homozygous locus including CDK5RAP2 in patients 1 and 2 and subsequent Sanger sequencing of CDK5RAP2 revealed a homozygous frameshift duplication of 2 bp in exon 7, leading to a premature stop codon (c.564_565dup, [p.(Lys189Argfs*15)] [GenBank: NM_018249.5] Chr9(GRCh37):g.123,298,747_123,298,748dup). Both parents were heterozygous carriers. Gene panel analysis was performed in patients 3–5. Patient 3 was found to carry ‘assumed compound-heterozygous’ CDK5RAP2 variants, as the parents were not available for testing: one 4 bp deletion in exon 4 (c.217_220del, [p.(Glu73Thrfs*4)] [GenBank: NM_018249.5]; Chr9(GRCh37):g.123313156_123313159del) and a 1 bp duplication in exon 36 (c.5495dup, [p.(Leu1832Phefs*3)] [GenBank: NM_018249.5]; Chr9(GRCh37):g.123156873dup). These two variants are predicted to induce a frameshift leading to null alleles. Patients 4 and 5 displayed a homozygous pathogenic mutation in exon 12. This single nucleotide duplication creates a premature stop codon 4 codons downstream (c.1303dup, [p.(Thr435Asnfs*4)] [GenBank: NM_018249.5]; Chr9(GRCh37):g.123280713dup) predicting a null allele. The father was heterozygous carrier (no sample available from the mother).
None of the mutations found in patients 1–3 are present in public databases (gnomAD, dbSNP, 1000 Genomes, the NHLBI Exome Variant Server and the Exome Aggregation Consortium (ExAC) browser). The variant identified in patients 4 and 5 has been reported only once, in the Latino population of the gnomAD database and has never been reported in the homozygous state.
Patients 6 and 7 were found to be compound heterozygous for truncating variants by trio whole-exome sequencing as recently described.7 Each patient carried one unreported variant and one with a very low minor allele frequency, both heterozygous, in the ExAC/gnomAD databases. Patient 6 harboured NM_018249.5(CDK5RAP2):c.[558_559delGA]+[c.4441C>T], p.[(Glu186Aspfs*32)]+[(Arg1481*)], Chr9(GRCh37):g.[123298753_123298754del];[123171568C>T] with the mother heterozygously carrying c.[558_559delGA]+[=], p.[(Glu186Aspfs*32)]+[=] (unreported) and the father heterozygously carrying c.[c.4441C>T]+[=], p.[(Arg1481*)]+[=] (rs587783390; ExAC: A=0.00005660/4, no homozygotes). Patient 7 harboured NM_018249.5(CDK5RAP2):c.[3928G>T];[4546G>T] p.[(Glu1310*)];[(Glu1516*)] Chr9(GRCh37):g.[123199600C>A];[123171463C>A] with the mother heterozygously carrying c.[4546G>T];[=] p.[(Glu1516*)];[(=)] (rs374351172; ExAC: A=0.00002472/3, no homozygotes, gnomAD: A=0.00002844/7, no homozygotes) and the father heterozygously carrying c.[3928G>T];[=] p.[(Glu1310*)];[(=)] (unreported).
Clinical characteristics
Clinical details are summarised in table 1. Microcephaly was confirmed at birth in all cases and was even detectable during the second or third trimester of pregnancy in five out of seven patients before reaching a mean occipitofrontal circumference (OFC) of −6±1.7 SD during infancy. Nevertheless, an unusual improvement of the OFC after age 2 for patients 1, 2 and 4 was noted (online supplementary figure 1). None of the patients had short stature beyond age 5. Areas of hypopigmentation or hyperpigmentation of the skin were systematically found in all patients. Motor development was also normal. Suspected around age 3 because of speech delays, the main functional consequence of brain volume reduction and simplified gyri, both obvious on brain MRI (online supplementary figure 2), was ID, ranging from mild to moderate (FSIQ from 40 (patient 5) to 67 (patient 1), see online supplementary figure 3). We cannot exclude the possibility that patient 5, with moderate ID may have a second hit related to the context of consanguinity. However, in all patients, a relative preservation of verbal comprehension (VCI 78 and 75 for patients 1 and 2) and visuospatial abilities (71 and 75 for patients 2, 3 and 6) as well as a preservation of mental manipulation was detected (78 and 73 for patients 1 and 4). None of the patients exhibited epilepsy.
Supplemental material
Supplemental material
Supplemental material
Developmental hypothalamic defect
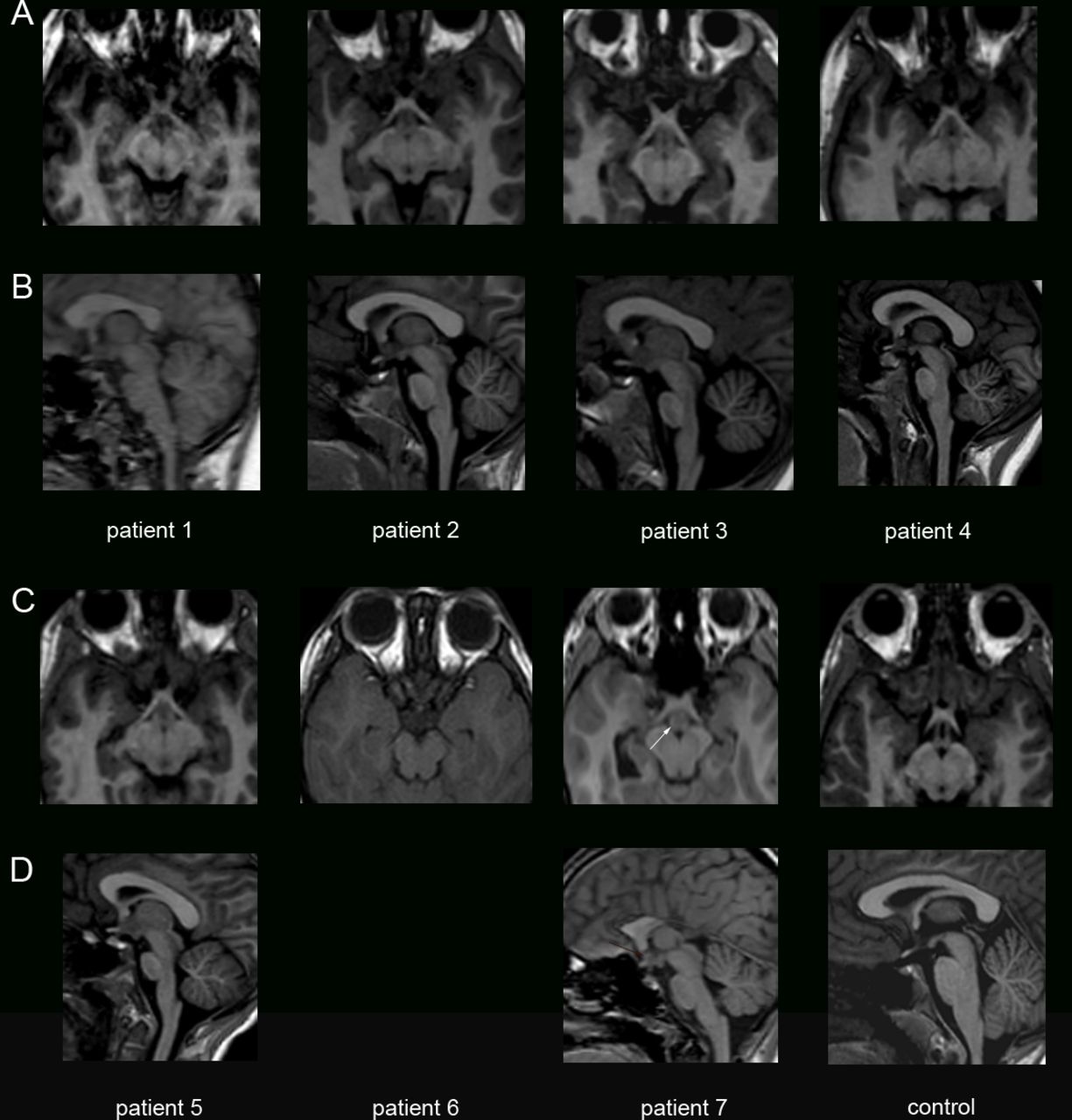
Patients 1, 2, 4, 5 and 7 displayed interhypothalamic adhesion (IHA), a grey matter band that connects the dorsomedial and ventromedial hypothalamic nuclei and/or arcuate, paraventricular or preoptic nuclei on both sides of the third ventricle32 (figure 2). IHA differs from hamartoma as it does not extend into the third ventricle. For patient 6, brain MRI was performed with another protocol including a series of 5 mm slices without T1-weighted 3D sequence, which did not allow to formally identify IHA. IHA was already noticed on the brain MRI performed at 1 year of age in patient 1, and was not enhanced after gadolinium injection, as classically described for glioma, another differential diagnosis of IHA. Taken together, these observations suggest that IHA, which is present at least in five out of seven patients, may be caused by a failure of the separation of hypothalamic nuclei at early developmental stages.

Developmental hypothalamic defects in patients with CDK5RAP2 variants. Coronal (A, C) and sagittal (B, D) T1-weighted images showing in patients 1, 2, 4, 5 and 7 a structure iso-intense to grey matter consistent with an interhypothalamic adhesion (IHA, white arrows), and absent in the healthy control. This grey matter band on coronal view and nodular on sagittal view is located at the level of the upper midbrain, closed to the anterior/inferior third ventricle. IHA is known to connect both dorsomedial and ventromedial hypothalamic nuclei and/or arcuate, paraventricular or pre-optic nuclei on both sides of the third ventricle.
Neurosensory impairment

Retinal pigmentary changes in patients with CDK5RAP2 variants. (A, B) Ocular fundus photography of patients 1 and 4 showing focal regions of hyperpigmentation and hypopigmentation in the retinal pigment epithelium (RPE). Note that hyperpigmented lesions appeared as small dark spots (patient 1, white arrows) or dark grey extended lesions (patient 4, black arrow) and were located in the macular zone and/or in the retinal periphery. Small round spots of hypopigmentation were scattered in the posterior pole (yellow arrows in patients 1 and 4). (C, D) Ocular fundus autofluorescence showing that hyperpigmented lesions appear as well-defined patches of hypoautofluorescence (figure 3C–D, white and black arrows), whereas hypopigmented spots appear as areas of hyperfluorescence, reminiscent of lipofuscin deposits/accumulation in the RPE in both patients (yellow arrows).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
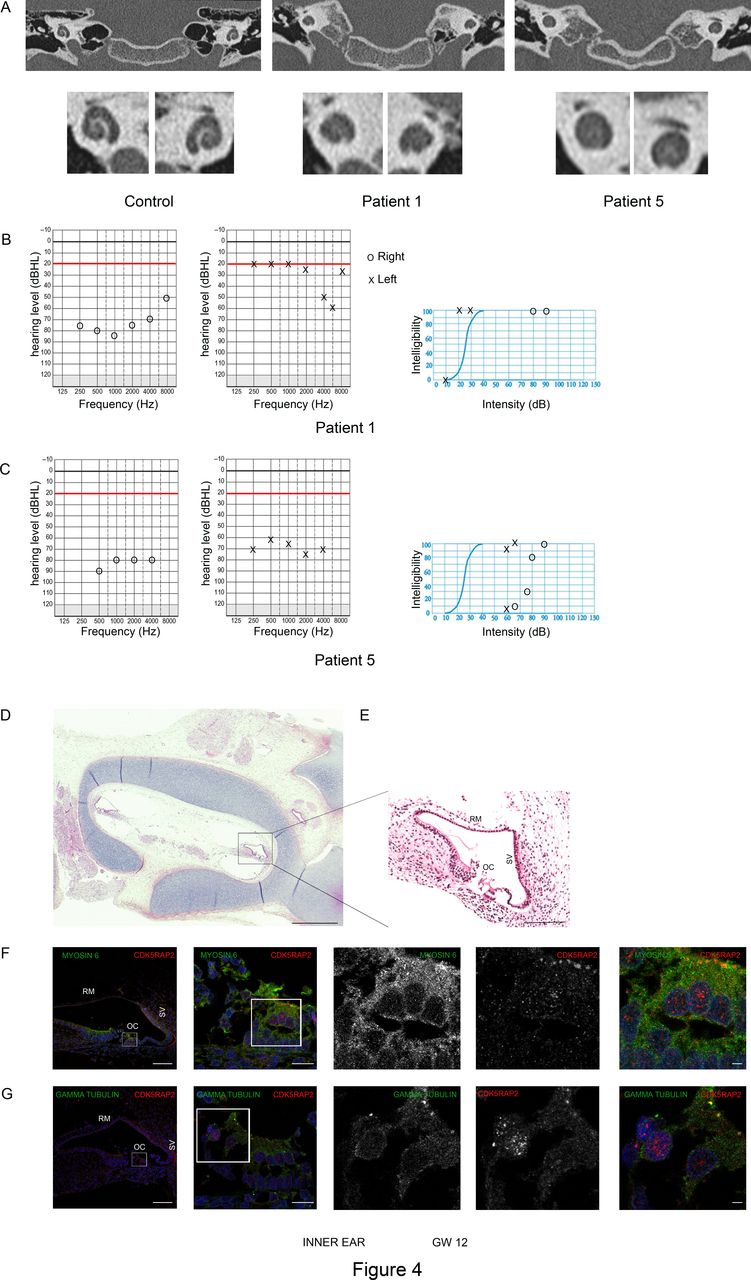
Congenital inner ear malformation responsible for progressive deafness in patients with CDK5RAP2 variants. (A) Temporal bone coronal CT scan images in patient 1 and 5 showing small cochlea with no visibility of the interscalar septa at the apical turn of the cochlea and a confluence of middle and apical turns of the cochlea, evocative of an incomplete partition type II according to Sennaroglu classification. (B, C) Tonal and vocal audiograms of patients 1 (B) and 5 (C). Circles and X symbolise joint air and bone-conducted hearing thresholds, respectively for right and left ears. (B) On the right ear, the ascending curve corresponds to a severe hearing loss whereas the left ear demonstrates a normal hearing threshold for conversational frequencies associated with a scotoma at 6000 Hz. (C) The mean hearing threshold of 80 dB on the right ear and 70 dB on the left ear indicate a bilateral hearing loss. (D, E) Horizontal section of temporal bone (D) of a control fetus at WG12 stained by H&E and high magnification on the cochlea (E) highlighting the structure of a normal fetal cochlea. RM, Reissler’s membrane; OC, organ of Corti; SV, stria vascularis. Scale bars: 25 mm (D), 500 µm (E). (F, G) Immunohistochemistry in the adjacent section of the cochlea of the control fetus at WG12 shown in (D). White squares indicate regions of the cochlea that are shown in higher magnification in the right part of the panel. (F) Co-labelling of CDK5RAP2 (red) and myosin VI (green, that stains the outer hair cells of the cochlea) and DAPI (blue) showing that CDK5RAP2 is expressed in myosin VI positive cells at the apical pole of outer hair cells. (G) Co-labelling of CDK5RAP2 (red) and gamma-tubulin (green, that stains the pericentriolar material at the centrosome) demonstrating that CDK5RAP2 is localised at the centrosome of the outer hair cells of the cochlea. Scale bars: 90 µm (first left panel), 15 µm (second left panel), 15 µm (right panel).
Microphthalmia and retinal pigmentation defects
Ophthalmological assessment showed normal biomicroscopy and no oculomotor disorder. The axial length was below the normal range in all patients (average: 20.9, from 20.3 to 22.3 mm; normal age >3 years: 23.5 mm) (tables 1 and 2, figures 3 and 4).
In all patients, examination of the ocular fundus revealed round or oval regions of focal hyperpigmentation or hypopigmentation scattered in the periphery and all along the vascular arcade. Hyperpigmented lesions consisted of either extended dark grey lesions or small dark lesions scattered in the macular zone and in the retinal periphery (figure 3A–B). Small round spots of hypopigmentation were either located in the macula (figure 3B) or scattered in the periphery (figure 3A–B). All hyperpigmented lesions were hypoautofluorescent (figure 3C–D), while hypopigmented spots were hyperautofluorescent (figure 3C–D). OCT was performed and found normal in patients 1 and 4.
All patients being hyperopic, best-corrected visual acuity was 20/20 except in the youngest patient (patient 3), aged 4 years and non-cooperative, whose visual acuity was superior to 20/40 in each eye. Scotopic and photopic flash ERGs as well as the EOG and pERG were similar to age-matched controls (data not shown).
Progressive hearing loss associated with congenital inner ear malformation
Patients 1, 3, 5 and 6 developed progressive unilateral or bilateral hearing loss, respectively diagnosed at age 9, 6, 7 and 10 (table 1). Audiometric evaluations at diagnosis showed severe hearing loss from 70 to 80 Hz in at least one ear for all four patients. Patient 1 had a unilateral scotoma of the left ear at 6000 Hz at 60 dB, and a 77.5 dB threshold in the right ear (figure 4B–C). Close to normal hearing levels were restored with conventional hearing aids. However, patient 5 did not receive early intervention/rehabilitation. This delayed medical support is likely to have increased the receptive and expressive speech impairment caused by PM itself and worsened ID. Surprisingly, although they had been stable for at least 4 years, patient 1 has recently undergone an improvement of his auditory thresholds, which is now at 42 dB in the right ear and 26 dB in the left ear on average, with a concordant vocal threshold.
As sensorineural hearing loss (SNHL) is very unusual in patients with PM, a temporal bone CT scan was performed for all patients except patient 6 (parental rejection), and revealed the same cochlear malformation in each case: small cochlea compared with normal values on CT33 and an absence of the interscalar septa at the apical turn (figure 4A and table 2), that is, compatible with the diagnosis of incomplete partition type II of the Sennaroglu classification.34 In addition, patient 2 displayed an enlarged vestibular aqueduct associated with this cochlear dysplasia. In all patients, the diameter of the bony cochlear nerve canal was reduced (<1.7 mm), in favour of a cochlear nerve hypoplasia.33
CDK5RAP2 detection in the fetal cochlear epithelium
The CDK5RAP2 protein is known to be expressed in the ventricular zone (VZ) of both mouse and human telencephalon throughout neurogenesis.16 However, its presence in the inner ear has not been documented so far. Using immunohistochemistry for CDK5RAP2 on human fetal tissues, we found that it is expressed in the developing cochlea in addition to the fetal brain. Co-labelling of CDK5RAP2 with γ-tubulin on human fetal brain sections at early (WG8) and mild (WG12) stages of neurogenesis confirmed that CDK5RAP2 is highly expressed at the centrosome of the apical radial glial cells (aRGCs) of the VZ (online supplementary figure 4). Immunolabeling on human fetal cochlea at WG12 showed that CDK5RAP2 is expressed in MYOSIN VI (a protein expressed in the cytoplasm of outer hair cells of the organ of Corti) positive cells (figure 4F (and Figure 4 D-E for low magnification). At the apical pole of these outer hair cells, CDK5RAP2 was also co-expressed with γ-tubulin (figure 4G) at the centrosome, concordant with its expression within the VZ. Thus, CDK5RAP2 is expressed early at the centrosome in the sensory epithelium of the organ of Corti as in the neuroepithelium.
Supplemental material
Discussion
In this study, we identify neurosensory impairments associated with the CDK5RAP2 microcephaly, including retinal pigmentation defects and a specific cochlear malformation responsible for progressive sensorineural hearing loss. All patients examined with our protocol displayed IHA, suggesting a failure in hypothalamic nuclei separation. CDK5RAP2 is thus crucial for neurosensory development in addition to brain development.
CDK5RAP2 and brain development
In agreement with the congenital defects observed, CDK5RAP2 is highly expressed during human neurocorticogenesis. Although the protein has been detected from WG18 onwards,16 we find that CDK5RAP2 is present as early as WG8 in the VZ of the human fetal cortex, at the centrosome of aRGCs. This highlights CDK5RAP2 requirement for early progenitors’ division and may explain why microcephaly is often detected antenatally. Similarly, aRGCs switch from symmetric (proliferative) to asymmetric (neurogenic) division in Cdk5rap2-deficient mice, which results in premature cell cycle exit, increased cell death and a reduced number of cortical neurons.15
Interestingly, OFC suddenly increased after age 2 in three patients (online supplementary figure 1), a phenomenon not previously reported in PM. The partial compensation observed could result from postnatal white matter development, myelination and/or synaptogenesis.35 36 Brain MRI early follow-up, including T2-weighted images and tractography should therefore be envisioned to assess myelination and synaptogenesis in CDK5RAP2 patients. Despite the reduction in neuron number, it is possible that neurons that have reached maturity manage developing a fairly efficient network, a situation that would be consistent with the mild ID observed, unlike patients with PM due toASPM mutations who have more pronounced ID.30 In agreement with this assumption, patients whose OFC increases after age 2 also showed a higher IQ. Larger cohorts would greatly help validating this hypothesis.
IHA was detected in five patients. This band of grey matter connects hypothalamic nuclei from the two sides of the third ventricle may be similar to a forme fruste of holoprosencephaly (HPE), which is restricted to the ventral part of the diencephalon and characterised by the non-separation of hypothalamic nuclei along the midline without fusion of the thalami. IHA is often associated with other CNS midline defects including corpus callosum dysgenesis, as seen in patients 6 and 7, or other defects not observed in our patients such as an absence of the septum pellucidum, optic pathway hypoplasia, hippocampal dysgenesis.32 37 However, IHA may also occur as an isolated asymptomatic feature.32 At their last examination, the children of our series did not exhibit any obvious endocrine disorder attributable to the hypothalamic defect: growth was within the normal range for weight and height; no diabetes insipidus or high blood pressure was noted. Although no micropenis was detected, tracking puberty onset would be relevant. Interestingly, patients 1 and 4 showed excessive weight gain (after age 8 and 9, respectively). During the second decade of life, we propose to assess energy metabolism in these patients.
PM and HPE have already been associated in patients carrying variants in the MCPH7 gene STIL.38 These patients exhibit a mild lobar form of HPE and PM with gyral simplification and interhemispheric cysts. This suggests that, as for STIL, CDK5RAP2 can be added to the list of genes causing a forme fruste of HPE in humans.
CDK5RAP2 and ear development
We show here that CDK5RAP2 loss of function affects the development of extracortical regions and results in a broader phenotype than just microcephaly. This observation is consistent with the pattern of expression of CDK5RAP2, which encompasses the cortical VZ and subventricular zones and the spiral organ of Corti, and indicates that this centrosomal protein is critical to the development and maintenance of neurosensory functions.
Malformations of the inner ear are found in 30%–40% of children with SNHL on imaging (CT, MRI).33 39 40 Incomplete partition of the cochlea type II34 refers to a cochlea that has a normal basal turn but a cystic apex with hypoplasia of the modiolus and absence of the interscalar septum. This malformation was obvious in all examined patients. Patient 2 also showed a dilated vestibule and vestibular aqueduct enlargement (Mondini dysplasia). Incomplete partition of the cochlea type II is a congenital cause of sensorineural hearing loss. This cochlear malformation typically occurs during WG7.41 Hearing loss may be mild to profound and must be evaluated with systematic audiometry. As CDK5RAP2 variants described in the present study have not been reported previously, one cannot rule out the possibility that the associated neurosensory impairments are variant-specific. However, hearing loss has been occasionally mentioned in previous CDK5RAP2 patients carrying other variants19 26 and our systematic study suggests that the neurosensory defects are likely constitutive of the CDK5RAP2 phenotype and may have been overlooked in previously reported cases.
The most common aetiology of Mondini dysplasia is a variation in the SLC26A4 gene involved in Pendred syndrome42 ,43 and in a non-syndromic hearing loss,44 but variants in >100 genes cause severe congenital or progressive hearing loss.45 Genes involved in genetic SNHL encode proteins playing a role in gene regulation, fluid homeostasis, synaptic transmission and hair cell and neuronal maturation, and in stereocilia function.45 To date, no interaction between such proteins and CDK5RAP2 has been reported. In humans, the cochlea develops between WG6–7 and 10–12, and the vestibule between WG9 and 13,41 that is, at the beginning of neurogenesis. It has been shown that the brain volume reduction related to Cdk5rap2-deficient mice is due to reduced neural progenitor proliferation, cell death and early neuronal differentiation during neurogenesis.15 It is thus possible that the observed simplification of the cochlear spiral also reflects defective proliferation or cell death leading to a depletion of the pool of progenitors and the early differentiation of the cochlear epithelium. Given that hearing loss was not congenital and was identified when the youngest patient was 6 years of age, we recommend that audiograms be performed systematically at the time of diagnosis, and possibly, repeated on a yearly basis. Neurosensory management of these patients is crucial as it considerably impacts communication, learning and integration abilities.
CDK5RAP2 and eye development
PM is not commonly associated with microphthalmia except in specific syndromes such as Warburg-Micro syndrome (#600118, RAB3GAP1, #614225, RAB3GAP2, #614222, RAB18, #615623, TBC1D20), microcephaly and chorioretinopathy type 3 (#616335, TUBGCP4 45) or type 6 (#251270, TUBGCP6 46) or microcephaly with or without chorioretinopathy, lymphoedema and mental retardation (#152950, KIF11 47). In the present study, we have shown that microphthalmia is a cardinal feature of the CDK5RAP2-associated phenotype, and confirms in humans what Zaqout et al have recently shown in Cdk5rap2 mutant mice (an/an).48 More surprising were the retinal pigmentation changes found in all the patients. Retinal hyperpigmentation could be related to skin hyperpigmentation spots. Hypopigmented spots that are hyperautofluorescent are reminiscent of lipofuscin deposits/accumulation in the retinal pigmentary epithelium (RPE). No functional consequences on visual acuity or ERG were noticed. OCT was normal for both patients tested, suggesting that lipofuscin accumulation does not alter the RPE-photoreceptor layer. Our patients still being children, their ophthalmological status obviously needs to be reassessed to check whether lipofuscin deposits are stable or increase with time and/or impact visual function. These retinal pigmentation changes do not resemble the well-characterised chorioretinopathies associated with microcephaly such as those caused by PLK4, TUBGCP6 (#25127046), TUBGC4 (#61633545) or KIF11 (#15295047) variants. We propose that this new entity, which resembles that described by Abdel-Saam et al prior to gene identification,49 may be related to CDK5RAP2 variants, because it is present in all patients of the present series.
Conclusion
Overall, our findings extend the phenotype associated with CDK5RAP2 microcephaly and indicate that MCPH3 is distinct from other MCPH by its association with neurosensory impairment. These results shed new light on the role of CDK5RAP2 in brain, ear and eye development and maturation.
Acknowledgments
The authors would like to thank all patients and their family. The authors would like to thank the CIC team (CIC 1426, Inserm) at Robert Debré Hospital in Paris who organised the families' visit and ensured the implementation of the clinical research protocol. The authors would also like to thank Dr Sowmyalakshmi Rasika for relevant comments and language editing during the preparation of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
LV and ME-B contributed equally.
Contributors HN analysed and interpreted all clinical data. LV and NT analysed the ocular and ENT phenotypes, respectively for all patients with the help of MM. ME analysed brain MRI and temporal bone CT scan for all patients. HN, LV, NT and ME helped writing the manuscript. AE performed the neuropsychological assessment for all patients. KS, AA, MLM, DH, YA and MZ were involved in patient recruitment and clinical management. SGC and FK conducted the research protocol. PL and FG performed the immunostaining in human tissues. VEG, AV and PG revised and edited the manuscript. SD and AR analysed and interpreted NGS data and performed segregation analysis. SP conceived the study, interpreted the results and wrote the manuscript.
Funding This work was supported by the Délégation à la Recherche Clinique et à l’Innovation de l’Assistance Publique Hopitaux de Paris, the Institut National pour la Santé et la Recherche Médicale (Inserm), the Université Paris 7, the Centre National de la Recherche Scientifique (CNRS), the DHU PROTECT, the Programme Hospitalier de Recherche Clinique (PHRC, grant agreement n° P100128/IDRCB: 2010-A01481-38), by ERA-NET grant 'Euromicro' (ANR-13-RARE-0007-01, ANR-16-CE16-0024-01 to SP, AV, VE, SD and PG and SNF 31ER30_154238 to AR) and radiz–Rare Disease Initiative Zurich, clinical research priority programme, University of Zurich.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The trial was approved by the National Ethics Committee (Comité de Protection des Personnes (CPP) Ile-de-France II).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Data used in the manuscript are available by request to the authors.