Article Text

Abstract
Background Rapid automatised naming (RAN) and rapid alternating stimulus (RAS) are reliable predictors of reading disability. The underlying biology of reading disability is poorly understood. However, the high correlation among RAN, RAS and reading could be attributable to shared genetic factors that contribute to common biological mechanisms.
Objective To identify shared genetic factors that contribute to RAN and RAS performance using a multivariate approach.
Methods We conducted a multivariate genome-wide association analysis of RAN Objects, RAN Letters and RAS Letters/Numbers in a sample of 1331 Hispanic American and African–American youth. Follow-up neuroimaging genetic analysis of cortical regions associated with reading ability in an independent sample and epigenetic examination of extant data predicting tissue-specific functionality in the brain were also conducted.
Results Genome-wide significant effects were observed at rs1555839 (p=4.03×10−8) and replicated in an independent sample of 318 children of European ancestry. Epigenetic analysis and chromatin state models of the implicated 70 kb region of 10q23.31 support active transcription of the gene RNLS in the brain, which encodes a catecholamine metabolising protein. Chromatin contact maps of adult hippocampal tissue indicate a potential enhancer–promoter interaction regulating RNLS expression. Neuroimaging genetic analysis in an independent, multiethnic sample (n=690) showed that rs1555839 is associated with structural variation in the right inferior parietal lobule.
Conclusion This study provides support for a novel trait locus at chromosome 10q23.31 and proposes a potential gene–brain–behaviour relationship for targeted future functional analysis to understand underlying biological mechanisms for reading disability.
- complex traits
- epigenetics
- genome-wide
- psychiatry
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Reading disability (RD, also known as developmental dyslexia) is a complex neurodevelopmental disorder characterised by difficulties in reading despite educational opportunity and normal intelligence, and is the most common neurodevelopmental disorder diagnosed in children ages 5–18 years, with a prevalence of 5%–17%.1 The genetic component of RD is strong, with family and twin-based studies estimating moderate to high heritabilities >0.50.2 To date, at least nine susceptibility loci for RD (DYX1-9) have been identified through linkage mapping and replicated in several independent populations, along with several candidate risk genes including KIAA0319, DCDC2 and DYX1C1.2 However, variants in these candidate genes and loci do not account for a substantial portion of the estimated heritability.3 In addition, genome-wide analyses predicated on quantitative measures of reading ability and case–control status for RD have identified novel variants but have had little success attaining genome-wide significance.4 This could be explained by low sample sizes in the studies, but another reason may be phenotypic heterogeneity of the RD phenotype.
One approach to studying the genetics of complex trait disorders such as RD is to examine relevant endophenotypes. An endophenotype is a quantitative trait measure that is correlated with a disorder or trait of interest due, in part, to shared (pleiotropic) underlying genetic influences. For a phenotype to be considered an endophenotype for a complex trait disorder, it must have a genetic component, be independent of clinical state (affected or unaffected), cosegregate with disorder status in a family and have reproducible measurements.5 Endophenotypes are conceived as being closer to the underlying biology than the corresponding complex disorder. This could improve the statistical power to identify genetic variants through larger effect sizes. Two potential endophenotypes for reading proficiency that satisfy these criteria are rapid automatised naming (RAN) and rapid alternating stimulus (RAS).
RAN tasks require sequential naming of visually presented, familiar items (eg, objects, letters, numbers or colours) as quickly and accurately as possible. RAN has been described as capturing a ‘mini-circuit’ of the reading network that taps into cognitive subprocesses critical for reading, such as automatic attentional processing.6 Reading and RAN performance are moderately correlated (0.28–0.57), with deficits in RAN performance observed in 60%–75% of individuals with RD.7–9 Furthermore, performance on RAN in kindergarten is predictive of later reading fluency and is stable through elementary school.10 11 RAN deficits even persist into adulthood.12 13
RAS tasks are similar in overall design to RAN tasks but involve sequential naming of items across alternating stimulus categories as quickly and accurately as possible (ie, alternating letters and numbers). RAS tasks evaluate the ability to direct attention while performing an automatic task like sequential naming.14 RAN and RAS are highly correlated (0.85), and like RAN, RAS performance can differentiate poor from typically developing readers and is highly predictive of later reading ability.14–16
RAN has a strong genetic component with heritability estimates ranging from 0.46 to 0.65.17 18 To date, several family-based studies have identified novel genetic loci linked to RAN and RAS performance, with some studies also supporting linkage to other previously identified DYX loci.19 The majority of studies focused on linkage mapping, while the limited number of association studies on RAN and RAS directly targeted the strongest candidate genes and/or variants identified for RD from the available literature. However, a recent genome-wide meta-analysis and multivariate analysis of RAN Digits, RAN Letters and RAN Pictures found significant associations with rs17663182 in chromosome 18q12.2 in a sample of 2562–3468 European individuals.20
Notably, genetic studies of RAN, RAS and RD were previously conducted on samples of largely European descent. This is problematic because for causal variants, differences in observed allele frequencies and linkage disequilibrium (LD) between populations could lead to associations observed in one population but not in another. Furthermore, lack of diversity in association studies leads to missed opportunities to identify novel trait loci present in certain populations, thus limiting the identification of biological clues surrounding the aetiology of a complex disorder.21 Using a single population to identify disease-associating regions constrains the ability to generalise relevant findings to other populations, and severely limits the benefits of biomedical research to only those populations with selected demographics.22 23
The present study aims to identify shared genetic factors that contribute to covariance across RAN Objects, RAN Letters and RAS Letters/Numbers using a multivariate genome-wide association study in 1331 unrelated children in the Genes, Reading, and Dyslexia (GRaD) study, a case–control sample of RD in Hispanic American and African–American youth. It builds on previous studies that investigated the genetic underpinnings of RAN and RAS performance as univariate traits. However, a univariate design can miss underlying covariance shared across two or more correlated traits and therefore has low sensitivity for detecting shared genetic factors.24 We hypothesise that the correlations between RAN and RAS performance can partially be attributed to shared genetic factors.17 To test this hypothesis, we apply a multivariate genetic analysis to leverage covariance across phenotypes and to increase statistical power to identify the presence of genetic pleiotropy across different RAN and RAS tasks.24
Methods
Subjects, recruitment and DNA collection
The GRaD study is a multisite, case–control study of RD in minority youth across the USA, Canada and Puerto Rico. Subject recruitment and collection of phenotype data and DNA were completed for 1432 unrelated children. Of these subjects, 1331 children who passed genotyping quality control were included in the analysis.
The Colorado Learning Disability Research Center (CLDRC) cohort is a family-based sample selected for RD, Attention-Deficit/Hyperactivity Disorder (ADHD) and other learning disabilities. DNA and phenotype data from 318 participants, one from each twinship/sibship, were randomly selected for analysis from a total of 749 participants.
The Pediatric Imaging, Neurocognition, and Genetics (PING) study is a cross-sectional sample of typically developing children ranging in age from 3 to 20 years old. Imaging genetics analysis was conducted on 690 subjects with complete phenotype and genotype data.
Detailed descriptions of phenotypes, recruitment, inclusion and exclusion criteria, self-reported demographic breakdown, and DNA collection for each of the three samples are reported in the online supplementary methods and tables S1–S2.
Supplemental material
Statistical analysis
A multivariate genome-wide association study (GWAS) of RAN Objects, RAN Letters and RAS Letters/Numbers was conducted using the R package MultiPhen.25 Joint models were corrected for the first three principal components (PC; online supplementary figure S1 and S2A) to correct for population stratification, sex, age and socioeconomic status (SES). A genome-wide significance threshold of p<5×10−8 was used to correct for multiple testing.26
Multivariate GWAS results were verified using a follow-up univariate GWAS on a latent variable composite of RAN Objects, RAN Letters and RAS Letters/Numbers in PLINK V.1.9 in the GRaD sample stratified by assignment to a ‘Hispanic American’ and ‘African American’ cluster based on genetic similarity to 1000 genomes reference populations AMR (Ad Mixed American) and ASW (African–Americans in the Southwest USA), respectively (online supplementary figure S3). For the Hispanic American cluster, the univariate GWAS model was corrected for the effects of age, sex, SES and the first three ethnic-specific PCs to correct for intraethnic stratification (online supplementary figure S2B). For the African–American cluster, the model was corrected for age, sex, SES and the first two ethnic-specific PCs (online supplementary figure S2C). A meta-analysis was then conducted in METAL using summary statistics from the latent naming speed GWAS.27 Univariate analysis of covariance (ANCOVA) testing the top SNP against measures of reading fluency (Test of Word Reading Efficiency (TOWRE) and Woodcock-Johnson 3 (WJ-III)) in the full GRaD sample was conducted in SPSS V.24. Models corrected for the effects of age, sex, SES and the first three genome-wide PCs (online supplementary figure S2A).
Replication was conducted using the CLDRC sample through joint analysis of RAN Colours, RAN Pictures, RAN Letters and RAN Numbers at SNPs identified in the GRaD discovery analyses. Models were corrected for the effects of age, sex and the first genome-wide PC (online supplementary table S2).
Detailed descriptions of statistical analyses are reported in the online supplementary methods.
Bioinformatic analysis
All analyses were conducted using positions mapping to genome build GRCh37/hg19.
Predicted tissue-specific functionality in the genome was estimated through the presence of well-characterised DNase 1 hypersensitivity sites and histone marks and using GenoSkyline.28 Precalculated, genome-wide, tissue-specific, posterior probability scores (GenoSkyline (GS) scores) for the whole brain and subregions of the brain (angular gyrus, prefrontal cortex, cingulate gyrus, anterior caudate, hippocampus, inferior temporal gyrus and substantia nigra) were obtained from the GenoSkyline database.
Follow-up analysis of epigenetic data from the Roadmap Epigenomics Project was conducted to identify predicted chromatin state in a genomic region of interest across specific tissue types. The 18-state model was evaluated in angular gyrus, prefrontal cortex, cingulate gyrus, anterior caudate, hippocampus, inferior temporal gyrus and substantia nigra. Data were visualised using the Washington University in St Louis EpiGenome Browser V.42.
The 3D Genome Browser was used to visualise publicly available chromatin contact maps at 10 kb resolution from adult hippocampal tissue.29 30
Detailed descriptions of the bioinformatic analysis are reported in the online supplementary methods.
Neuroimaging genetic analysis
Neuroimaging genetics analysis in the PING sample was conducted using a linear regression association test in PLINK V.1.9. All analyses were corrected for the effects of age, sex, handedness, scanner device,31 intracranial volume, highest parental education, family income and the first four genome-wide PCs to correct for population stratification (online supplementary figure S2E). Left and right cortical volumes across seven regions of interest associated with the canonical reading network were tested for genetic association (online supplementary table S3). The left and right hippocampus was also included based on follow-up analyses, for a total of 16 assessed regions.
Results
Multivariate and univariate associations for RAN and RAS
There are significant Pearson correlations across RAN Objects, RAN Letters and RAS Letters/Numbers in the GRaD sample ranging from 0.621 to 0.781 (p<1×10−140; online supplementary table S4), providing the rationale to use a multivariate GWAS. Multivariate GWAS of RAS Letter/Numbers, RAN Objects and RAN Letters reveals a genome-wide significant effect for rs1555839 (p=4.03×10−8; genomic inflation factor [λ]=1.047; figure 1, online supplementary figure S4, table 1) located approximately 5 kb upstream from ribosomal protein L7 pseudogene 34 (RPL7P34). RPL7P34 is a long non-coding RNA (lncRNA) on chromosome 10 mapping between the genes LIPF and LIPJ. Additional SNPs approaching significance (p<1×10−5) cluster within a 70 kb region of chromosome 10q23.31 that spans RPL7P34 and a gene called renalase (RNLS; MIM: 609360) (figure 1B). To determine whether specific variables may be driving the multivariate signal, we conducted an independent, post-hoc, univariate examination of RAN Objects, RAN Letters and RAS Letters/Numbers on the top SNPs identified in the multivariate analysis. The independent analysis shows that the SNPs located on chromosome 10q23.31 have an effect across all RAN and RAS tasks (table 1, online supplementary figure S5–S7). The strongest association in the chromosome 10q23.31 region is with SNP rs1555839 (β=−0.31, p=1.71×10−9) for RAS Letters/Numbers.

(A) Manhattan plot summarising the results of the multivariate genome-wide association study (GWAS) for RAN Objects, RAN Letters and RAS Letters/Numbers and (B) LocusZoom plot of genomic region surrounding genome-wide significant SNP, rs1555839. The genome-wide-associated SNP, rs1555839, is marked by a black diamond. lncRNA pseudogene, RPL7P34 (chr10: 90,377,980–90,378,691), is not represented in the LocusZoom plot. Grey line: Bonferroni correction for multiple testing, p<5×10−8. lncRNA, long non-coding RNA; RAN, rapid automatised naming.
Multivariate genome-wide association study results
To further validate the findings from the multivariate GWAS of the full GRaD cohort, we performed a GWAS meta-analysis of Hispanic American (AMR cluster; n=883) and African–American (ASW cluster; n=441) participants in the GRaD sample on a latent naming speed variable (online supplementary figure S3). rs1555839 and rs6963842, previously implicated in the multivariate GWAS of RAN Objects, RAN Letters and RAS Letters/Numbers, show suggestive association (p<1×10−6; λ=1.013; online supplementary figure S8, table S5). rs701825 on chromosome 10, also implicated in the discovery multivariate GWAS, is also highly associated with naming speed latent variable in the meta-analysis. However, the significant heterogeneity statistic indicates a difference in the magnitude of the effect between Hispanic Americans and African–Americans on naming speed, but the direction of effect remains the same (online supplementary table S5). Both the multivariate GWAS and the latent naming speed meta-analysis implicate the same regions of chromosome 7q31.1 and 10q23.31, providing independent evidence that findings are not from hidden population stratification.
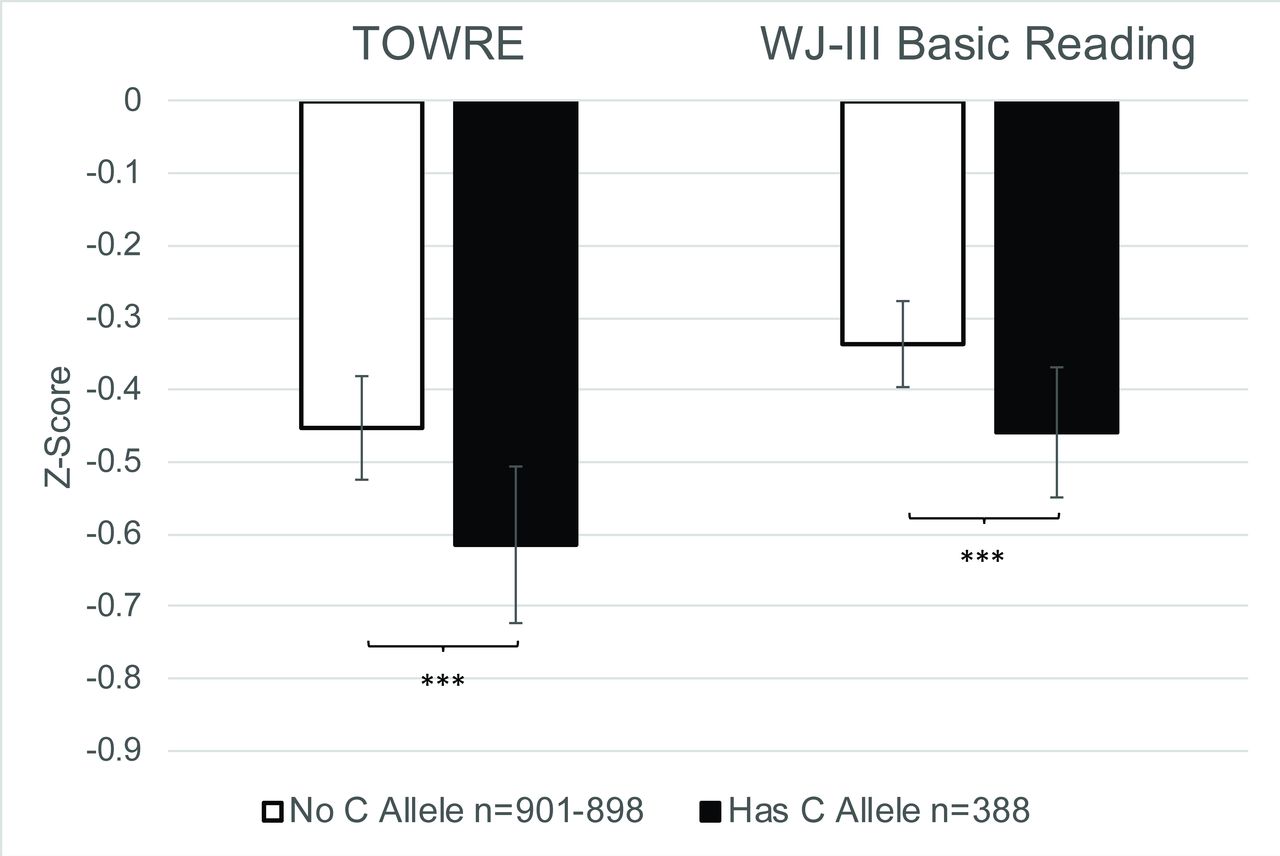
Due to significant correlations observed between RAN/RAS tasks and measures of word reading (r=0.325–578, p<1×10−32; online supplementary table S4), we evaluated whether allelic variation at rs1555839 was associated with mean differences in performance for TOWRE and WJ-III Basic Reading. Univariate ANCOVA shows a significant effect of rs1555839 with TOWRE (F[1,1281]=12.50, p=4.21×10−4, ηp 2=0.01) and WJ-III Basic Reading (F[1,1278]=13.47, p=2.53×10−4, ηp 2=0.01) (figure 2). Overall, performance on reading fluency and word reading is worse in the presence of the C allele at rs1555839, which is the same allele and direction of effect associated with reduced scores on RAN and RAS.

Mean differences in TOWRE and WJ-III Basic Reading performance by allelic variation at rs1555839 in the GRaD study. In the presence of the C allele, there is a significant reduction in TOWRE and WJ-III Basic Reading performance, while correcting for the effects of age, sex and population stratification (the first three principal components). Error bars represent 95% CI. TOWRE n=901, WJ-III n=898; ***p<0.001. GRaD, Genes, Reading, and Dyslexia study.
Replication of multivariate GWAS and latent naming speed GWAS meta-analysis
Next, we brought forward all three SNPs that achieved genome-wide significance or were suggestive in both the discovery multivariate GWAS and the latent naming speed GWAS meta-analysis for replication in the CLDRC. Assessments of RAN Colours, RAN Letters, RAN Pictures and RAN Numbers were available in 318 unrelated participants. RAN Objects and RAS Letters/Numbers were not assessed in the CLDRC. Multivariate analyses of RAN Colours, RAN Letters, RAN Pictures and RAN Numbers show a significant replication of rs701825 on chromosome 10, while we observe marginal significance with rs1555839. rs6963842 on chromosome 7 does not replicate (table 2). Post-hoc univariate association analysis shows a significant replication of rs1555839 and rs701825 for RAN Letters (table 2, online supplementary table S6). The direction of effect on RAN Letters is the same for both cohorts (online supplementary table S6).
CLDRC multivariate replication
Bioinformatic analysis
RNLS, a gene located within 50 kB of rs1555839, is expressed in all regions of the brain (Genotype-Tissue Expression (GTEx) median >1 transcripts per million (TPM))32 and throughout the human lifespan (BrainSpan).33 LIPJ is expressed at low levels in the cerebellar hemisphere and cerebellum (median <0.7 TPM). LIPF is not expressed in the brain in either the GTEx or BrainSpan databases.
We used GenoSkyline to evaluate the presence of predicted functional regions of the genome surrounding the 70 kb region of chromosome 10q23.31 spanning rs2576167–rs701825 implicated in the discovery multivariate GWAS using epigenomic annotations from the National Institutes of Health Roadmap Epigenomics Project and Encyclopedia of DNA Elements (ENCODE). Within all sampled tissues, GenoSkyline predicts tissue-specific functionality surrounding our top SNPs. The first major peak (chr10:90341871–90343706) encompasses the promoter region of RNLS, reaching the maximum GS score of 1 for all tissues (table 3, online supplementary figure S9). The second major peak (chr10: 90376471–90376828) is within 2 kB of RPL7P34, reaching a maximum GS score of 1 in the whole brain, 0.886 in the hippocampus, 0.629 in the anterior caudate and 0.880 in the cingulate gyrus (figure 3A, online supplementary figure S9). GS scores for the angular gyrus, dorsolateral prefrontal cortex, inferior temporal gyrus and substantia nigra are less than 0.5 (online supplementary figure S9).
Association results of rs1555839 tested against reading-related regions of interest in the PING study

{kind=link}
{kind=link}
{kind=link}
Epigenetic examination of chromosome 10q23.31 containing the top-performing SNPs in the GRaD discovery analysis. (A) Plot of GS scores obtained from GenoSkyline indicating the posterior probability for functionality in the brain at each genomic locus. A GenoSkyline (GS) score of 1 indicates a high probability for functionality, while a GS score of 0 suggests no functionality. (B) 18-state chromatin model from Roadmap Epigenomics Project showing predicted chromatin states based on the presence of H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3 and H3K27ac sampled across different regions of the brain. (C) Location of genes implicated in the GRaD discovery analysis, and location of linkage disequilibrium blocks (black bars) in CEU, AMR and YRI populations in 1000 Genome Project Phase 3. (D) Chromatin contact map of adult hippocampal tissue showing regions of chromatin interactions at 10 kB resolution. Open stars represent the position of top chromosome 10q23.31 SNPs in the GRaD multivariate genome-wide association study (GWAS) analysis. AMR, Ad Mixed American; GRaD, Genes, Reading, and Dyslexia study.
To determine the chromatin state of the 70 kb region of chromosome 10q23.31 that contributes to high posterior probability scores for tissue-specific functionality in the genome, we evaluated the 18-state model previously generated by the Roadmap Epigenomics Project. The region closely associated with the most significant SNPs in the GRaD discovery analysis (rs10749593, rs7913742, rs1555839) flanks an active enhancer site in the hippocampus and cingulate gyrus. These SNPs also flank regions of heterochromatin containing ZNF genes and repeats in the cingulate gyrus and inferior temporal lobe (figure 3B). In addition, an active transcription start site for RNLS is predicted in all brain regions (figure 3B).
Examination of LD blocks across different ethnic groups sampled in 1000 Genomes Phase 3 that are represented in the GRaD sample (Utah Residents with Northern and Western European Ancestry (CEU), Ad Mixed American (AMR) and Yoruba in Ibadan, Nigeria (YRI)) shows a single LD block spanning approximately 39 kb encompassing the segment that spans rs2576167–rs7913742 but not including rs1555839 or rs701825 (figure 3C; online supplementary table S7). However, in CEU and AMR, a single LD block spans the 70 kb segment that spans all SNPs of interest (figure 3C; online supplementary table S7). Values for D’ for CEU, AMR and YRI populations range from 1 to 0.87, indicating high correlation across these SNPs in chromosome 10q23.31.34 35 Taken together, these data suggest a similar genetic architecture across ethnic populations within this region.
To evaluate whether a potential enhancer–promoter interaction exists between the predicted enhancer site near RPL7P34 and active transcription start site for RNLS in the hippocampus, we examined extant Hi-C contact maps publicly available through the 3D Genome Browser.29 30 There is evidence that a chromatin interaction is present between a region containing the predicted enhancer site and the active transcription start site at a 10 kb resolution in the adult hippocampus (figure 3D). No extant Hi-C contact maps are publicly available for the cingulate gyrus to evaluate a potential enhancer–promoter interaction in this region.
PING neuroimaging genetic analysis
Following replication of rs1555839 and epigenetic analysis showing predicted functionality in brain tissue within the region of chromosome 10q23.31, we conducted a targeted neuroimaging genetics analysis in 690 typically developing children of various ancestral backgrounds from the PING sample. The C allele of rs1555839 is associated with lower cortical volume in the right inferior parietal cortex, after correcting for multiple testing (β=−437, p=2.43×10−3; table 3). There is a suggestive association between rs1555839 and lower cortical volumes in the left hippocampus (β=−53.64, p=9.42×10−3; table 3).
Discussion
The present study is one of the first to examine the genetics of reading-related traits in an admixed population of Hispanic American and African–American children. Here, we describe a region of chromosome 10q23.31 that exceeds genome-wide significance with pleiotropic effects across RAN Objects, RAN Letters and RAS Letters/Numbers—highly correlated tasks that are predictive of later reading outcome and reading ability in children and adults.6 We validated the GWAS results with a genome-wide meta-analysis of a univariate latent naming speed variable that implicates the same region of chromosome 10. Although results from the genome-wide meta-analysis did not attain genome-wide significance, this could reflect the loss of power by stratifying the GRaD cohort into smaller sample sizes, or perhaps the loss of power to identify genetic factors with pleiotropic effects using a univariate approach.24
Multivariate replication in a European sample further supports a shared effect from a small segment of chromosome 10 across RAN tasks. Within the CLDRC sample, only one endophenotype, RAN Letters, overlapped with the GRaD sample in the present analysis. Although this is not a perfect replication of the primary analysis, the high correlation across different RAN and RAS tasks16 17 and the underlying hypothesis of the overall study to identify shared genetic factors contributing to general RAN and RAS performance justify replicating in the CLDRC sample. Furthermore, the significant and same direction of effect we see with chromosome 10q23.31 SNPs, rs1555839 and rs701825, on RAN Letters in a different ethnic group suggests that these findings can be generalised across more than one population. This is supported by LD maps showing a similar underlying genetic architecture of this region across representative 1000 Genomes Project populations in CEU and AMR, as well as the high correlation across SNPs in CEU, AMR and YRI.
The most highly associated SNP in the RAS/RAN genome-wide screen, rs1555839, is located 5 kb upstream from the lncRNA, RPL7P34. The function of RPL7P34 is unknown, and the roles of lncRNAs in the genome are poorly understood. lncRNAs form a class of non-protein coding transcripts over 200 nucleotide bases long, and have characteristics that suggest functionality including tissue-specific expression, regulated expression, and regulation of gene expression and their networks.36 lncRNAs are reported to recruit transcription factors and interact with chromatin modifiers, suggesting that they facilitate epigenetic regulation of the genome.37 Approximately 40% of lncRNAs are expressed in the brain and are hypothesised to play critical roles in neural development such as neural proliferation and differentiation.38
Epigenetic examination of the region surrounding RPL7P34 shows a predicted active enhancer site based on the presence of H3K4me1 and H3K27ac histone modifiers specifically in the hippocampus and cingulate gyrus. It is possible that RPL7P34 plays a role in the recruitment of proteins that bind to enhancer sites, which promote transcription of nearby genes. The nearby gene RNLS, approximately 30 kb away, encodes the closest predicted transcription start site within the brain. Examination of chromatin contact maps shows that RPL7P34 and the promoter region of RNLS interact in the hippocampus, suggesting a potential enhancer–promoter interaction. Taken together, these results suggest that the enhancer region downstream from RPL7P34 could regulate RNLS. However, further targeted functional analyses to experimentally validate the molecular and epigenetic function of RPL7P34 and the nearby enhancer site would have to be conducted. It is also important to note that the epigenetic and Hi-C data in the brain analysed by the Roadmap Epigenomics Project were obtained from only two individuals.39 Although these data offer some clues to potential functionality, they should be interpreted with caution, considering the number of subjects sampled in the Roadmap Epigenomics Project, and lack of substantiation by a cell-based assay directly showing enhancer function in this region.
RNLS, implicated in our GWAS and epigenetic analysis, encodes a flavin adenine dinucleotide-dependent amine oxidase, called renalase, which metabolises catecholamines such as norepinephrine and dopamine.40 41 Renalase is known to be secreted by the kidneys, circulates in blood, and modulates cardiac function and blood pressure.41 Gene expression data from GTEx and BrainSpan also suggest that renalase is present in the human central nervous system, including the cortex, hippocampus and cingulate gyrus.40 In vitro analysis of monoamine oxidase activity of renalase shows that it is most efficient in metabolising dopamine, but is also effective in metabolising epinephrine and norepinephrine.41 Two major neuromodulatory systems in the brain, dopaminergic and noradrenergic, play important functions in motivation, attention, learning and memory formation—especially in the hippocampus and cingulate gyrus—but the functional role of renalase in metabolising neurotransmitters in the brain is currently unclear.40 42 However, genetic variants in RNLS have been associated with schizophrenia, a neuropsychiatric disorder that may be caused by an imbalance in neurotransmission. Specifically, a recent study showed that human-induced pluripotent stem cell (hiPSC)-derived neurons from patients with schizophrenia had altered catecholamine release relative to control hiPSC-derived neurons.43
This is the first study to implicate RNLS in reading-related domains. However, it is not the first gene associated with the metabolism of catecholamines. COMT encodes catechol-O-methyltransferase, which also degrades catecholamines in the brain, and has been associated with variation in reading-related tasks as well as functional networks associated with reading ability.44 45 There is also evidence showing neurochemical differences between poor readers and typically developing controls, suggesting that there could be alterations in how neurotransmitters are metabolised in reading impaired individuals.46
Neuroimaging-genetic analysis indicates that rs1555839 is associated with variation in cortical volume in the right inferior parietal cortex. Limited studies have been conducted on structural neuroanatomical correlates of RAN, but there is evidence that RAN performance is associated with grey matter volumes in bilateral occipital-temporal and parietal-frontal regions, which include the right inferior parietal cortex.47 In addition, functional variation in the right inferior parietal cortex has also been implicated in several reading-related tasks and RD.48
The small sample size is a limitation of this study for GWAS analyses. However, based on power calculations for a multivariate GWAS, we had modest power of 0.68 to identify a genetic variant with a minor allele frequency of 0.16, effect size ranging from 0.03 to 0.013, and cross-phenotype correlation of 0.7, in a sample with 1263 individuals (both phenotype and genotype data) and α=5×10−8—all parameters applicable to this study and the observed SNPs showing most significant associations.49 While hidden population admixture could be confounding our analyses, the calculated genomic inflation factor is within acceptable ranges (λ<1.05) after correction, suggesting adequate control for population stratification. Furthermore, we split the GRaD cohort by genetic ancestry using k-means clustering with AMR and ASW reference populations and conducted a genome-wide meta-analysis on a latent naming speed variable derived from performance across RAN Objects, RAN Letters and RAS Letters/Numbers, and we replicated the findings in an independent sample of European ancestry, attaining similar results across all modes of analysis.
In conclusion, by leveraging different data sources and types across neuroimaging and epigenetic data, we identified and replicated association of a region of chromosome 10q23.31 with pleiotropic effects across RAN, RAS and reading abilities in a sample of Hispanic American and African–American youth. There is converging evidence that non-coding regions of the genome have an impact on reading-related traits, and the identification of an lncRNA associated with a reading endophenotype lends additional support.50 The association of RNLS also reinforces the hypothesis that alterations in neurochemical modulation in the brain could contribute to impairments in reading performance. However, further functional assays must be conducted to shed mechanistic light on the pathways involved.
This study highlights the importance of studying the genetic architecture of RD across diverse ethnicities and how genetic effects and variants may differ (or be similar) across populations. This is critical in our understanding of the biological mechanisms that contribute to RD, and is necessary for presymptomatic identification and development of precision intervention strategies that are relevant to all populations.
Electronic database information
1000 Genomes Project (http://www.internationalgenome.org/).
GenoSkyline (http://genocanyon.med.yale.edu/GenoSkyline).
National Institutes of Health Roadmap Epigenomics Project (http://egg2.wustl.edu/roadmap/web_portal/index.html).
3D Genome Browser (http://promoter.bx.psu.edu/hi-c/view.php).
Pediatric Imaging, Neurocognition, and Genetics Portal (http://ping.chd.ucsd.edu/).
FreeSurfer (http://surfer.nmr.mgh.harvard.edu/).
LDlink (https://analysistools.nci.nih.gov/LDlink/?tab=home).
Acknowledgments
We extend our sincere gratitude to all the individuals and their families who participated in the GRaD, CLDRC and PING studies. We are also grateful to the research assistants for their help with recruitment and data collection. We thank the staff at the Yale Center for Genome Analysis for genotyping services. We also thank Dr Mellissa DeMille, Dr Andrew DeWan, Dr Jeffrey Malins, Dr Chintan Mehta and Dr Hongyu Zhao for invaluable discussion and critical evaluation of this manuscript.
References
Footnotes
Contributors DTT designed the experiments, performed statistical analyses and wrote the manuscript. AKA and SP provided computational and bioinformatic support and edited the manuscript. JCF performed statistical analyses and edited the manuscript. RB, DEH, MWL, EMM, EGW, MW, JCD, AG, CF, SEF, RKO, BFP, SDS and JB-H collected data and edited the manuscript. JRG obtained funding for the GRaD study, collected data and edited the manuscript.
Funding The Genes, Reading, and Dyslexia study was funded by The Manton Foundation (JRG, JCF, JB-H, DTT). The Colorado Learning Disabilities Cohort was funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development National Institutes of Health Grant (P50HD027802; JRG, JCD, RKO, BFP, SDS, EGW). The Pediatric Imaging, Neurocognition, and Genetics study was funded by the National Institute on Drug Abuse and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (RC2DA029475). DTT was funded by the Lambert Family and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (T32HD07094 and K99HD094902). AKA was funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (T32HD007149). The Max Planck Society supported AG, CF and SEF, and funded the genetic analyses of the CLDRC cohort. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Summary statistics will be made available through the NHGRI-EBI GWAS Catalog https://www.ebi.ac.uk/gwas/downloads/summary-statistics