Article Text

Abstract
Background Cryptorchidism or failure of testicular descent is the most common genitourinary birth defect in males. While both the insulin-like peptide 3 (INSL3) and its receptor, relaxin family peptide receptor 2 (RXFP2), have been demonstrated to control testicular descent in mice, their link to human cryptorchidism is weak, with no clear cause–effect demonstrated.
Objective To identify the genetic cause of a case of familial cryptorchidism.
Methods We recruited a family in which four boys had isolated bilateral cryptorchidism. A fourth-degree consanguineous union in the family was reported. Whole exome sequencing was carried out for the four affected boys and their parents, and variants that segregated with the disorder and had a link to testis development/descent were analysed. Functional analysis of a RXFP2 variant in cell culture included receptor localisation, ligand binding and cyclic AMP (cAMP) pathway activation.
Results Genomic analysis revealed a homozygous missense variant in the RXFP2 gene (c.1496G>A .p.Gly499Glu) in all four affected boys and heterozygous in both parents. No other variant with a link to testis biology was found. The RXFP2 variant is rare in genomic databases and predicted to be damaging. It has not been previously reported. Functional analysis demonstrated that the variant protein had poor cell surface expression and failed to bind INSL3 or respond to the ligand with cAMP signalling.
Conclusion This is the first reported genomic analysis of a family with multiple individuals affected with cryptorchidism. It demonstrates that recessive variants in the RXFP2 gene underlie familial cryptorchidism and solidifies the link between this gene and testicular descent in humans.
- cryptorchidism
- undescended testis
- RXFP2
- exome sequencing
- familial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Cryptorchidism (OMIM 219050) or failure of testicular descent is the most common genitourinary birth defect in males, found in between 1.6% and 9.0% of boys.1 It can affect one (unilateral) or both (bilateral) testes and can be isolated or associated with additional anomalies. Cryptorchidism, if left untreated, can cause serious complications such as testicular cancer or infertility.2 Testicular descent from an intra-abdominal position into the scrotum is often described as having two main phases: the transabdominal phase and the inguinoscrotal phase. This descent is guided by the gubernaculum (the rudder of the testis), a ligament-like structure derived from the primitive mesenchyme.3 During the transabdominal phase, the hypertrophy and growth of the gubernaculum steers the testis to the caudal part of abdomen. The descent of the testis requires hormonal factors produced by the fetal testis itself, such as INSL3 and androgens (see Mäkelä et al 4 for a review). Several genes and pathways have been implicated in testicular descent and cryptorchidism, mainly from work in mouse models.4 This includes the insulin-like peptide 3 (INSL3) hormone and its receptor, relaxin family peptide receptor 2 (RXFP2), which are essential for transabdominal migration of the testis in the mouse.5–10 These genes are expressed in Leydig cells,11 from where it is thought that the INSL3 hormone dilates the gubernaculum and allows the opening of the inguinal canal during the transabdominal descent phase.3
While mutations in these genes cause cryptorchidism in rodent models, the evidence for them directly causing cryptorchidism in humans is weak, and few studies have convincingly shown cause–effect, with most studies only suggesting association. The INSL3 receptor RXFP2, also known as leucine‐rich repeat‐containing G protein‐coupled receptor 8 (LGR8) or G-protein-coupled receptor affecting testis descent (GREAT), is located at chromosome 13 and encodes a protein with an extracellular ligand‐binding domain, seven transmembrane domains and one intracellular domain.12 When mutated in mice, this gene causes intra-abdominal bilateral cryptorchidism.8 10 Evidence for its involvement in humans is scant, and just one variant, p.Thr222Pro, has been described in patients with consistent functional evidence. In 2002, Golov and colleagues published a single patient carrying this heterozygous variant,8 and further research suggested that this variant was over-represented in Italian patients with cryptorchidism and not in controls.13 Functional analysis suggested that this variant has compromised activity.13 Since then, with the increased availability of genomic sequencing and large genetic variation databases, the association of this variant with cryptorchidism has been called into question. Of note, this is the only variant in RXFP2 present in ClinVar and it is now classified as a variant of uncertain significance. Indeed, this variant was reclassified based on a study in 2010 in which the authors screened for the RXFP2 p.Thr222Pro variant in 577 Spanish men (187 with cryptorchidism and 390 controls) and in 550 Italian men (199 with cryptorchidism and 351 controls) and found that in the Spanish study population, the variant was present at similar frequency in both cases (3 of 187; 1.6%) and controls (7 of 390; 1.8%) while in the Italian study population, the variant was more frequent in cases (9 of 199; 4.5%) than in controls (5 of 351; 1.4%), with an OR of 3.17 (p=0.031).14 It was concluded that p.Thr222Pro is a frequent variant in the Spanish population with no pathogenic effect, although the variant seemed to confer a mild risk for cryptorchidism in the Italian population.14 Several other studies in different populations have shown no enrichment of this variant in patients with cryptorchidism compared with controls.15 Indeed, the genome aggregation database (gnomAD) lists this variant as having a minor allele frequency of 0.004403, and homozygous variants are described. Taken together these data exclude a clear-cut cause–effect relationship between the p.Thr222Pro variant and cryptorchidism.
One roadblock to finding definitive genetic causes of cryptorchidism is the lack of convincing familial cases in which genetics have been applied. Only a handful of publications have described comprehensive familial cases of cryptorchidism,16–20 with none conclusively identifying a genetic cause. In summary, since its initial description, the exact role of RXFP2 in human cryptorchidism has remained elusive, with no clear cause–effect yet demonstrated.
Here we carried out exome sequencing analysis in a family in which four boys had familial bilateral cryptorchidism and found a rare recessive missense variant in RXFP2. This provides the first rigorous human genetic evidence that RXFP2 variants directly cause cryptorchidism. It also implies that RXFP2 has an autosomal recessive mode of inheritance in familial cases, rather than the previously suggested autosomal dominant mode.
Materials and methods
Patient recruitment and consent
Patients were recruited as part of their clinical care at Paediatric Endocrinology Services at Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, India. The family gave written informed consent for conducting detailed genetic testing from DNA of both parents and four boys, and for publishing, under the PGIMER ethics board.
Whole exome sequencing
DNA was extracted from EDTA blood from all four affected children and both parents. Whole exome library preparation and sequencing was performed by the Australian Genomics Research Facility using Agilent SureSelect Human All Exon V6 (Agilent Technologies, Santa Clara, California, USA) on a NovaSeq 6000 Sequencing System (Illumina, San Diego, California, USA), respectively. The sequencing data were analysed using cpipe,21 which is based on the Broad Institute’s best practice for variant calling. The mean coverage obtained for the six patient samples was 101 bp (95% CI 71.9 to 130) and the variants were deposited into SeqR for further analysis (https://seqr.broadinstitute.org/).
Cell culture and transfection
Human embryonic kidney (HEK) 293 T cells used for RXFP2 and RXFP2 Gly499Glu expression were maintained in complete medium (Dulbecco’s Modified Eagle Medium) supplemented with 10% fetal bovine serum, 1% L-glutamine and 1% penicillin/streptomycin. Cells were cultured in 175 cm2 flasks in incubators maintained at 37°C, with 5% CO2 and 85% humidity. For functional assays, cells were first seeded into 24-well or 96-well plates and transfected the following day with receptor DNA using LipofectAMINE 2000 (Invitrogen).
Mutagenesis
The mutation of glycine-499 to glutamic acid in human RXFP2 was achieved by QuikChange mutagenesis with PrimeStar polymerase (Takara Clontech) on N-terminally FLAG-tagged human RXFP2 in pcDNA3.1 according to manufacturer’s instructions and similarly to previously described.22 Notably, FLAG tagging of the receptor does not affect ligand binding or activation.23 The sequence of pcDNA3.1-FLAG-RXFP2 Gly499Glu was confirmed by DNA sequencing on both strands (Centre of Translational Pathology, University of Melbourne) to ensure that there were no additional mutations.
FLAG receptor expression assays
Cell surface and total cellular expression of FLAG-tagged RXFP2 receptors expressed in HEK293T cells was measured using a method described previously.24 Twenty-four hours after transfection in 24 well plates, cells were washed once in assay buffer (Tris buffered saline pH 7.4, 2 mM CaCl2) and fixed for 15 min by addition of assay buffer containing 3.7% formaldehyde (for cell surface) or 3.7% formaldehyde/0.25% Triton-X (for total cell expression). Cells were then washed twice with assay buffer, blocked for 45 min in assay buffer containing 1% bovine serum albumin, incubated at room temperature with mouse anti-FLAG M1 monoclonal antibody (Sigma Aldrich), washed once in assay buffer, incubated at room temperature in goat anti-mouse Alexa 488 conjugated antibody (Invitrogen) and washed twice in assay buffer. Finally, cells were lysed and transferred to black walled 96-well optiplates for fluorescence measurement at 520 nm after excitation at 479–491 nm on a Polarstar Omega plate reader (BMG Labtech). Non-specific background was determined using cells transfected with empty vector and FLAG-RXFP2 Gly499Glu expression was expressed as the percentage of the FLAG-RXFP2 receptor expression. Data are presented as mean±SD of data normalised to RXFP2 expression from at least eight independent experiments each performed in triplicate. Raw cell surface and total expression data for FLAG-RXFP2 Gly499Glu was tested against FLAG-RXFP2 for statistical significance in GraphPad PRISM 8 using paired t-tests.
Eu3+-labelled INSL3 whole cell binding assay
FLAG-RXFP2 Gly499Glu was tested for its ability to bind INSL3 in comparison to FLAG-RXFP2 using a Europium (Eu3+)-labelled INSL3 (Eu-INSL3) whole cell saturation binding assay as previously described.25 Non-specific binding was determined by addition of 1 µM INSL3 and fluorescence was read on an Omega POLARstar plate reader using a time-resolved fluorescence protocol with excitation at 340 nm and emission at 614 nm. Data from at least three independent experiments, all performed in triplicate were pooled and presented as mean fluorescent specific binding±SEM using GraphPad Prism 8. Experiments were further analysed by nonlinear regression one-site binding curves to determine the K d value for FLAG-RXFP2. Cyclic AMP (cAMP) reporter gene assay cAMP activity in response to INSL3 of HEK293T cells expressing FLAG-RXFP2 Gly499Glu or FLAG-RXFP2 receptors was measured using a pCRE β-gal reporter gene assay as described in detail previously.26 Cells were incubated with increasing concentrations of INSL3 at 37°C for 6 hour, after which media was aspirated and the cells frozen at −80°C overnight. Forskolin (5 µM) was used as a positive control as it directly activates adenylate cyclase to increase intracellular cAMP. The amount of cAMP-driven β-gal expression was determined in cell lysates as described.26 Experiments were performed in triplicate at least three times, and data were pooled and presented as percentages of the response induced by 5 µM forskolin. Data were fit to a three-parameter sigmoidal dose–response curve using GraphPad Prism 8.
Results
Familial cryptorchidism affecting four boys in a single family
In October 2017, four boys from a single family (aged 9–15 years), with congenital bilateral cryptorchidism presented to the paediatric surgery department of our tertiary care paediatrics centre in North India (figure 1A). They were referred to the paediatric endocrinology services for endocrine evaluation. The boys were born out of a fourth-degree consanguineous marriage in a Muslim family. The father and a fifth male child were unaffected (figure 1A). All affected boys underwent circumcision in infancy (for religious faith) and all were noted to have bilateral undescended testes from early infancy. There was no history of hyperpigmentation, bifid scrotum or hypospadias. Also, there was no history of any urinary complaints or growth failure. None of the males in the extended family had similar history or infertility. All the boys had undergone one-sided orchidopexy at local hospital (around 1 year before presentation to our centre). On examination, the three boys of pubertal age were progressing well with spontaneous onset of puberty. All the boys had normal penile growth and scrotal development for their age and all the boys had one testis palpable in the scrotum (on the side orchidopexy was performed). In the elder boys (Sibs 1 and 2), the surgically fixed palpable testis was smaller in size compared with the two younger brothers. This was likely due to these older boys having had a relatively more delayed orchidopexy procedure (at age 14 and 13.5 years, respectively) and testicular tissue degeneration. The details of genital examinations are provided in table 1. The clinical findings of testicular size and location were confirmed by ultrasonography and MRI of the pelvic and inguinoscrotal region. Physical growth parameters were normal for all the boys. There was no history of bone problems (deformities or fractures) or other significant medical history.

A variant in relaxin family peptide receptor 2 (RXFP2) is associated with familial cryptorchidism. (A) Family pedigree. The proband and siblings are indicated by black squares. The grandfather had normal testes as per history and the youngest sibling was examined and genitalia was found to normal. (B) IGV screenshot of the RXFP2 c.1496G>A.p.Gly499Glu variant found as homozygous in all four affected boys and heterozygous in both parents.
Summary of the clinical details and results of laboratory evaluation of the four boys (Sibs)
The gonadal hormonal profile and testicular function was assessed with beta-human chorionic gonadotropin (HCG) stimulation. The results of endocrine hormonal assessment are summarised in table 1. Serum luteinising hormone (LH) and follicle stimulating hormone (FSH) levels were in pubertal range for all except youngest affected child (aged 9 years). Baseline androgen levels were consistent with the pubertal stage of the boys. Serum anti-Müllerian hormone (AMH) was also expectedly low in the older boys who are in puberty. The beta-HCG stimulated testosterone (T), dihydrotestosterone (DHT) and their ratio (T:DHT) were normal for all the boys (table 1).
In October to November 2017, three elder sibs underwent laparoscopy to localise the undescended testis and orchidopexy was performed successfully with both testes fixed in their normal physiological position in the scrotum. On last follow-up 1 year after the surgery (November 2018), Sib 1 had left testis 2–3 cc and right testis 15 cc; Sib 2 had left testis 12 cc and right testis 12–15 cc; Sib 3 had left testis 8 cc and right testis 10–12 cc. Sib 4 was still waiting diagnostic laparoscopy and orchidopexy for the right testis at the last follow-up.
Genetic analysis
Given the clinical profile and hormonal results, we had discounted the possibility of a disorder of androgen synthesis or action, as there were no features of undervirilisation and puberty had set in spontaneously. For example, a normal T:DHT ratio as well as a lack of hypospadias/micropenis suggested it was not 5-alpha reductase deficiency. We thus considered that this familial cryptorchidism may be due to a genetic mutation in the INSL3 gene or its receptor RXFP2, or in a novel causative gene. Therefore, to take an unbiased approach we decided to carry out whole exome sequencing for this family. Given the inheritance, we postulated that the familial cryptorchidism could be caused by a recessive variant, a maternally inherited X-linked variant, or a maternally inherited autosomal dominant variant. It was also possible that a dominant variant causative in all four boys was mosaic in the mother or father. We therefore first looked for rare variants with any of these patterns of inheritance in genes previously known to be involved in DSD/testicular development. Just one gene carried a variant: RXFP2.
RXFP2.c.1496G>A.p.Gly499Glu
All four affected boys were homozygous for a G>A change at genome position chr13:32366935, which is predicted to cause a glycine to glutamic acid at amino acid 499. This variant had good read depth and quality (figure 1B). In Exome Aggregation Consortium (ExAC), this variant has a minor allele frequency of 4.119e-05, being only found in South Asia (MAF of 0.0003028), where five people carried heterozygous alleles. GnomAD frequency in South Asia is 0.0002613. No homozygous cases are reported in ExAC or gnomAD. This amino acid is highly conserved and lies within the third transmembrane domain (figure 2A,B). The amino acid change predicted to be damaging by SIFT and probably damaging by PolyPhen. Following this, we also looked for any recessive gene variants that were not on our ‘DSD or testicular development’ list. This revealed a second gene carrying a variant of uncertain significance: ZC3H13.

The relaxin family peptide receptor 2 (RXFP2) Gly499Glu variant affects a highly conserved residue in the transmembrane domain. (A) Homology model of the RXFP2 receptor showing the location of our patient variant (Gly499Glu) and the previously implicated variant, Tyr222Pro. Receptor domains are highlighted; low density lipoprotein class A (LDLa) module in magenta, leucine-rich repeats (LRRs) in orange and transmembrane domain (TMD) in green; TM3 is blue, with residue G499 in yellow and T222 in purple. (B) Enlarged model of the RXFP2 TMD showing the position of the mutant G499E and orientation of glutamic acid side chain in yellow. Homology models for RXFP2 TMD (residues 411–747) and LRR domain (residues 109–389) were constructed using Swiss-Model with no constraints and the RXFP2 LDLa structure was available (PDB: 2M96). Images were prepared in Pymol molecular graphics system. (C) Glycine 499 is highly conserved from humans to fish.
ZC3H13:c.4113_4118delGGACAG.p.Asp1376_Arg1377del
All four patients also carried a homozygous in-frame deletion in the ZC3H13 gene at chr13:46541841, which was heterozygous in both parents (online supplementary figure 1A). ZC3H13 is a regulator of RNA m6A methylation and plays a role in splicing.27 This gene has not been previously associated with any human phenotypes, although it has been linked with colorectal cancer.28 The variant is present in gnomAD at a frequency of 0.002582 in South Asia with 79 heterozygotes and one homozygote reported. The deletion falls within a 27 bp region of DNA which encodes a repeated array of nine consecutive arginine (five) and aspartic acid (four) residues (online supplementary figure 1B, C); therefore, annotation of the deletion may vary. Of note, this region is not well conserved with two or more Arg/Asp pairs of amino acids missing in many other species (online supplementary figure 1C). This region does not fall within any annotated protein domains. ZC3H13 is only ~14 Mb from the RXFP2 gene so it appears the two variants may be segregating together in this consanguineous family explaining why all four affected boys have homozygous changes in both genes.
Supplemental material
Variants with X-linked or autosomal dominant inheritance
No variants with X-linked inheritance were found, although several (15) rare (<1×10−3 in ExAC and 1000G) maternally inherited heterozygous variants were found in all four boys . Just three of these genes are represented in OMIMand none are associated with any disorder involving the gonads or sexual reproduction.
The RXFP2 variant is non-functional in vitro
Given its essential role in testicular descent in the mouse and its previous, although weak, association to cryptorchidism in humans, the RXFP2 variant seems the most likely causative variant. This is supported by the fact that the additional variants have no link to testicular development or disease in mammals. We therefore wanted to test whether the RXFP2 variant caused a loss of activity in the protein which could explain the patients’ phenotype.
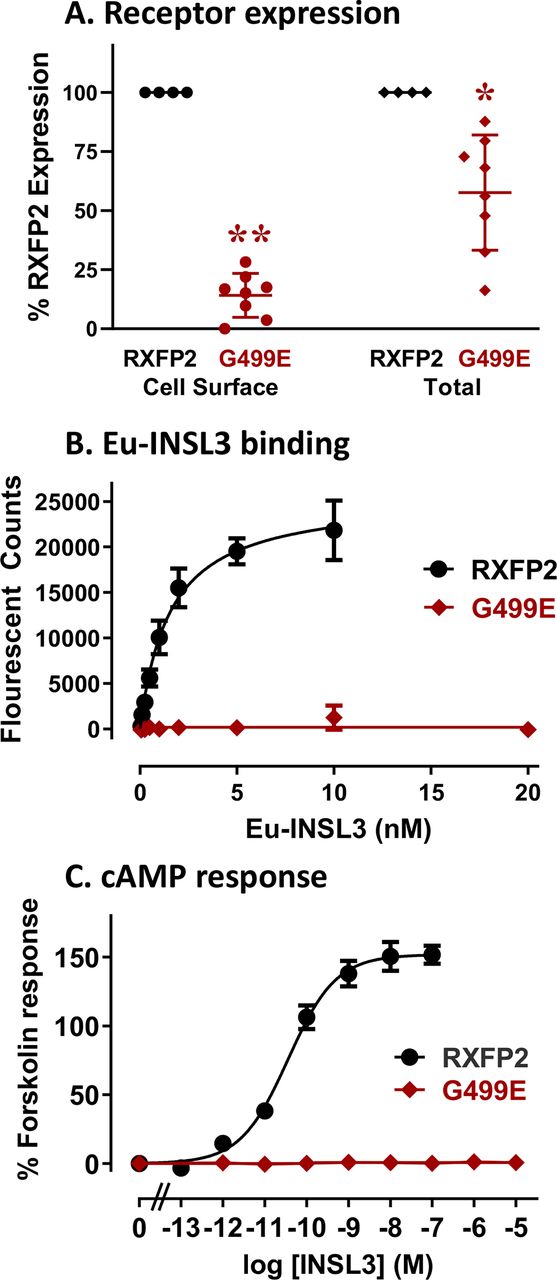
The affected amino acid, Gly499, is positioned in the third transmembrane domain of RXFP2 (figure 2A,B) and is highly conserved across species (figure 2C). To assess whether the glycine to glutamic acid change at amino acid position 499 affects the function of the RXFP2 protein, we performed mutagenesis on a well characterised pcDNA3.1-FLAG-RXFP2 construct7 to generate pcDNA3.1-FLAG-RXFP2 Gly499Glu (G499E). The construct was transfected into HEK-293T cells and tested in parallel with pcDNA3.1-FLAG-RXFP2 for both cell surface and total expression measured using the FLAG epitope, binding of Eu-INSL3 and cAMP activity responses to INSL3. FLAG expression assays demonstrated that the Gly499Glu variant was poorly expressed on the surface of HEK-293T cells and also showed lower total protein expression (figure 3A; p<0.01 and p<0.05, respectively). Eu-INSL3 binding assays and cAMP activity assays in response to INSL3 highlighted the high affinity and potency of INSL3 at RXFP2 but clearly showed that the small amounts of Gly499Glu variant at the cell surface were non-functional (figure 3B,C). These assays suggest that this genetic variant would cause a loss of activity in vivo.

{kind=link}
{kind=link}
{kind=link}
The relaxin family peptide receptor 2 (RXFP2) Gly499Glu variant is non-functional when expressed in cells. (A) Cell surface and total expression of FLAG-RXFP2 Gly499Glu (G499E; red) compared with wild type (WT) FLAG-RXFP2 (black) in human embryonic kidney-293T (HEK293T) cells as a percentage of expression. (B) Whole cell Europium (Eu)-insulin-like peptide 3 (INSL3) saturation binding assays comparing INSL3 binding of the Gly499Glu variant (red) compared with WT RXFP2 (black). (C) Cyclic AMP (cAMP) activity induced by INSL3 in HEK-293T cells expressing the Gly499Glu variant (red) compared with WT RXFP2 (black). (*p<0.05, **p<0.01).
Discussion
In this report, we thoroughly characterise a family in which four boys are affected by bilateral cryptorchidism. Cryptorchidism can often be part of an undervirilisation phenotype in which testicular or hormonal activity is disrupted (such as in 46,XY Disorders of Sex Development). Indeed, both 5-alpha reductase deficiency and androgen insensitivity syndromes, the two most common causes of undervirilisation, can run in families. In the family described here, these two possibilities were less likely given the phenotype of isolated undescended testes with no additional features of genital ambiguity/under-virilisation. Further, the spontaneous onset of puberty in three boys in pubertal age without any gynaecomastia excluded the above differential diagnoses, as well as hypogonadotrophic hypogonadism. Consistent with this, genetic analysis uncovered just one variant in a gene associated with a reproductive disorder, RXFP2.
Of interest, we found that serum FSH was abnormally high in the four affected boys (and increased over time) suggesting testicular (Sertoli cell) degeneration with age or with increasing duration of undescended testes. Serum AMH was low in the pubertal boys (normal) but may suggest Sertoli cell degeneration as well. The boys (especially in the two elder siblings) need to be assessed regarding potential for fertility. We plan to conduct semen analysis and sperm counts and mobility in the follow-up.
While RXFP2 has also been linked to osteoporosis in adult humans and RXFP2-deficient mice show reduced bone mass,29 no evidence for any bone anomalies or defects were found in this family, although this will also be monitored.
While cryptorchidism is common, very few large pedigrees affected by this have been described in detail. Indeed, we believe this is just the second study in which a family with numerous affected individuals has had genetic analysis and the first to have utilised an unbiased sequencing approach such as WES. In 2009, Feng and colleagues published a family with a proband, two cousins and maternal grandfather all with unilateral undescended testis. Targeted sequencing of the two exons of INSL3 and exons 8, 14, 17 of RFXP2, identified a heterozygous INSL3.p.Val39Gly substitution present in proband, mother and mosaic in the grandmother. It was not found in the affected cousins or grandfather, however, and showed no loss of activity in in vitro assays.19 We proposed that an unbiased sequencing approach be applied in large pedigrees in the future.
Our genetic analysis of the four affected boys and their parents found a homozygous missense variant in the RXFP2 gene, causing a Gly499Glu change within the third transmembrane domain. This variant is rare and predicted damaging. Interestingly, due to the consanguinity within the family reported here, it appears that the four affected boys have inherited a tract of DNA on chromosome 13, which includes the RXFP2 variant as well as an additional variant of uncertain significance in the ZC3H13 gene. Indeed, 43 other variants (synonymous and/or common) with the same inheritance are found within this region. It is always possible that this variant or other changes in this region are playing a role, although we did not find evidence for this and our in vitro assays strongly suggest that the RXFP2 variant is a loss of function variant. Indeed, our functional testing found that the variant protein expressed very poorly at the cell surface and what protein was expressed was not able to bind ligand or demonstrate any response to INSL3 with cAMP signalling. These tests indicate this missense variant is a null. Notably, Gly499 is positioned in the third transmembrane domain of RXFP2 in a position, 3.29 (Ballesteros and Weinstein numbering), which is involved in ligand binding of other Class A G-protein-coupled receptors (GPCRs).30 The substitution with glutamic acid in the mutant protein results in a large charged side chain which may project into the conserved GPCR Class A binding pocket and interfere with ligand binding and/or the conformation flexibility required for ligand-mediated activation. This disruption could also result in perturbed folding of the protein in the endoplasmic reticulum resulting in reduced total receptor expression and limited processing and trafficking of the receptor via the Golgi apparatus to the cell surface. Interestingly, just one other RXFP2 variant linked to cryptorchidism in some populations has been shown to have a loss of activity. Unlike our variant, this controversial heterozygous Thr222Pro variant is present in the protein’s ectodomain. Like our variant, functional analysis of Thr222Pro indicated reduced receptor surface expression of the variant protein13 and reduced relaxin-induced signal transduction (cAMP) compared with WT, however, when the variant protein was co-transfected with WT protein (to mimic its heterozygous format in patients), no reduction in activity was observed.8
To conclude, this study is the first to demonstrate a clear pathogenic variant (with loss of functional activity) in a known gonadal descent gene, which shows perfect inheritance/segregation in a family with multiple affected individuals. It is also the first report of a homozygous variant in the RXFP2 gene. We therefore conclude that recessive inheritance of variants in the RXFP2 gene are a cause of familial cryptorchidism.
Acknowledgments
We would like to thank the patients and their families for their participation and support of our study. We would also like to thank Tania Ferraro and Sharon Layfield for technical assistance at the Florey Institute.
References
Footnotes
KA and RK contributed equally.
Contributors KA carried out genomic analysis and coordinated the study. RK was the treating clinician and compiled all clinical notes. GR handled DNA and carried out genomic analysis. KB carried out bioinformatic analysis. SB carried out variant functional testing under supervision of RAB. MAM was the treating surgeon and contributed to clinical notes. AS oversaw the study. KA, RK, GR and RAB co-wrote the manuscript.
Funding Genomics work and GR are supported by National Health and Medical Research Council of Australia (NHMRC) program grant [1074258]. KLA is supported by an NHMRC project grant [1156942]. AHS is supported by a NHMRC fellowship [1154187]. Research at the Florey was supported by NHMRC project grants [1100676] and [1043750] and the Victorian Government Operational Infrastructure Support Program. RADB is supported by an NHMRC Research Fellowship [1135837].
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available upon reasonable request.