Article Text

Abstract
Background This study evaluates 6 years of prenatal rasopathy testing in the Netherlands, updates on previous data and gives recommendations for prenatal rasopathy testing.
Methods 424 fetal samples, sent in for prenatal rasopathy testing in 2011–2016, were collected. Cohort 1 included 231 samples that were sequenced for 1–5 rasopathy genes. Cohort 2 included 193 samples that were analysed with a 14-gene next generation sequencing (NGS) panel. For all mutation-positive samples in both cohorts, the referring physician provided detailed ultrasound findings and postnatal follow-up. For 168 mutation-negative samples in cohort 2, solely clinical information on the requisition form was collected.
Results In total, 40 (likely) pathogenic variants were detected (9.4%). All fetuses showed a variable degree of involvement of prenatal findings: increased nuchal translucency (NT)/cystic hygroma, distended jugular lymph sacs (JLS), hydrops fetalis, polyhydramnios, pleural effusion, ascites, cardiac defects and renal anomalies. An increased NT was the most common finding. Eight fetuses showed solely an increased NT/cystic hygroma, which were all larger than 5.5 mm. Ascites and renal anomalies appeared to be poor predictors of pathogenic outcome.
Conclusion Fetuses with a rasopathy show in general multiple ultrasound findings. The larger the NT and the longer it persists, the more likely it is to find a pathogenic variant. Rasopathy testing is recommended when the fetus shows an isolated increased NT ≥5.0 mm or when NT of ≥3.5 mm and at least one of the following ultrasound anomalies is present: distended JLS, hydrops fetalis, polyhydramnios, pleural effusion, ascites, cardiac defects and renal anomalies.
- rasopathy
- prenatal
- ngs
- increased nt
- noonan syndrome
Statistics from Altmetric.com
Introduction
Noonan syndrome (MIM: 163950) is characterised by postnatal short stature, distinctive facial features, congenital heart defects, variable degree of developmental delay and other structural abnormalities.1 The incidence is 1 in 1000–2500 live births.2–4 Noonan syndrome and phenotypically overlapping syndromes such as Costello syndrome (MIM: 218040) and cardiofaciocutaneous syndrome (MIM: 115150) are part of the so-called rasopathies and are caused by at least 16 genes (BRAF (MIM: 164757), CBL (MIM: 165360), HRAS (MIM: 190020), KRAS (MIM: 190070), LZTR1 (MIM: 600574), MAP2K1 (MIM: 176872), MAP2K2 (MIM: 601263), NRAS (MIM: 164790), PPP1CB (MIM: 600590), PTPN11 (MIM: 176876), RAF1 (MIM: 164760), RIT1 (MIM: 609591), SHOC2 (MIM: 602775), SOS1 (MIM: 182530), SOS2 (MIM: 601247) and SPRED1 (MIM: 609291)), the PTPN11 gene being the most prevalent gene.5 6 Prenatal features of rasopathies have long been documented and can include increased nuchal translucency (NT) and/or cystic hygroma, distended jugular lymph sacs (JLS), hydrops fetalis, polyhydramnios, pleural effusion, ascites, cardiac defects and renal anomalies.7–12 It has been previously estimated that mutations in the rasopathy genes are found in 6.7%–19% of fetuses with increased NT and additional anomalies on ultrasound.10 13 To date, only five studies have systematically evaluated the prenatal phenotype of Noonan syndrome.10 12 14–16
Croonen et al 10 performed a detailed study of prenatal diagnostic Noonan syndrome testing in fetuses with abnormal ultrasound findings (including enlarged NT above the 95th percentile, cystic hygroma, distended JLS, ascites, hydrops fetalis, pleural effusion, polyhydramnios, congenital heart disease and renal abnormalities) and showed a 17.3% mutation detection rate. The aim of Croonen’s study was to provide a protocol for prenatal Noonan syndrome testing.10 However, only 1–4 genes (KRAS, PTPN11, RAF1, and SOS1) were tested in their diagnostic study group (fetus with normal karyotype and specific abnormal ultrasound findings) and 10 genes (BRAF, HRAS, KRAS, MAP2K1, MAP2K2, NRAS, PTPN11, RAF1, SHOC2 and SOS1) in their anonymised study group (fetus with normal karyotype and increased NT, cystic hygroma, fetal hydrops and/or cardiac anomalies). Currently, a next generation sequencing (NGS) gene panel of 14 genes is used in the Netherlands in order to detect mutations in both prenatal and postnatal cases suspected of a rasopathy. Recently, a new guideline with regard to prenatal testing of a rasopathy in the presence of an increased NT was implemented in the Netherlands17; therefore, we considered it of clinical relevance to update on the previously published data.
Additionally, this study focuses on expanding the knowledge of the prenatal phenotype of a rasopathy. We performed a retrospective study in a consecutive series of 193 fetuses at time of diagnostic testing suspected of a rasopathy and another consecutive series of 231 fetuses in which a prenatal NGS panel of 14 genes involved in rasopathies was performed.
Methods
Patients
Included in this study is a consecutive series of 424 prenatally sampled fetuses with one or more of the following ultrasound findings: increased NT/cystic hygroma (defined as NT ≥3.5 mm), distended JLS, pleural effusion, ascites, polyhydramnios, cardiac defects and/or renal anomalies and a normal chromosomal microarray result. Cystic hygroma is considered an outdated term and combined with increased NT in this study.18 The samples were received from the Clinical Genetics departments of all University Medical Centers in the Netherlands and analysed in the Genome diagnostic laboratory of the Radboud University Medical Center in Nijmegen between January 2011 and December 2016. Of these 424 samples, 231 were received and analysed between January 2011 and September 2014 (cohort 1) and 193 between October 2014 and December 2016 (cohort 2). Samples were sent in as cultured or uncultured amniotic fluid sample, chorionic villi sample or DNA extracted from amniotic fluid or chorionic villi samples. In a few samples, DNA from the umbilical cord or fetal fibroblasts after termination of pregnancy was sent in.
Referring clinical geneticists of the mutation positive samples were asked to provide additional details on the clinical prenatal phenotype and postnatal phenotype if available. The mutation negative samples from cohort 2 were analysed only by the clinical information provided on the requisition form by the referring doctor.
DNA analysis
In cohort 1 (231 samples), 1–5 most commonly described rasopathy genes (BRAF, KRAS, MAP2K1, PTPN11 and/or RAF1) were parallel sequenced. All detected (likely) pathogenic variants or variants of uncertain significance were subsequently tested in parents if parental DNA was available.
Cohort 2, consisting of 193 fetal samples, was tested using the NGS rasopathy gene panel. The coding sequences and splice sites of A2ML1 (MIM: 610627), BRAF, CBL, HRAS, KRAS, MAP2K1, MAP2K2, NRAS, PTPN11, RAF1, RIT1, SHOC2, SOS1 and SPRED1 were sequenced by ion semiconductor sequencing (Ion AmpliSeqTM Noonan panel, ThermoFisher Scientific) combined with Sanger sequencing. All detected (likely) pathogenic variants or variants of uncertain significance were subsequently confirmed by Sanger sequencing and tested in parents if parental samples were available. Five class variant classification system was used: clearly not pathogenic variant (Class 1), unlikely pathogenic variant (Class 2), variant of uncertain clinical significance (Class 3), likely pathogenic variant (Class 4) and clearly pathogenic variant (Class 5).19
Statistical analysis
The data were analysed in SPSS V.22. For statistical analysis, a χ2 test, independent t-test, Shapiro-Wilk test, Mann-Whitney U test, logistic regression and descriptive statistics were used. A probability value of less than 0.05 was considered significant.
Results
Total
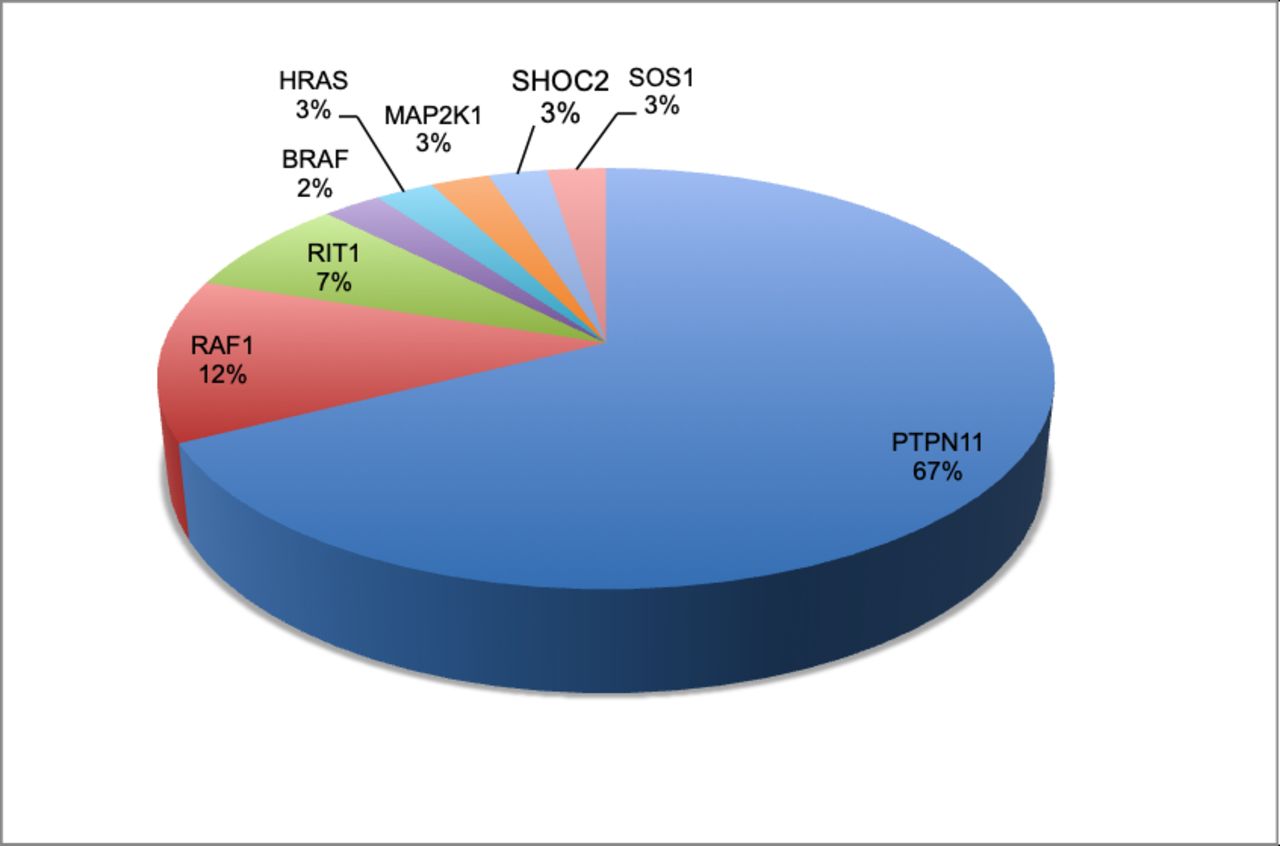
Over the 6-year period (2011–2016), 44 out of 424 fetuses were found to have a (likely) pathogenic variant or variant of unknown significance in one of the 14 rasopathy genes (10.4%). Forty variants had either been described before and shown to be pathogenic (Class 5) or were likely pathogenic (Class 4) (9.4%). Four variants were first considered variants of unknown significance (Class 3), but after segregation analysis, they were considered as likely benign variants (Class 2) as they were parentally inherited and the parent did not show a rasopathy phenotype. These variants were found in the RAF1, RIT1, SOS1 and HRAS genes. Twenty-seven of the 40 (likely) pathogenic variants were found in the PTPN11 gene (67.5%). Five fetuses showed a (likely) pathogenic RAF1 gene variant (12.5%). Three pathogenic variants were seen in the RIT1 gene (7.5%). In the BRAF, HRAS, MAP2K1, SHOC2 and SOS1 genes, a (likely) pathogenic variant was found in one fetus each (2.5% each gene) (figure 1). Thirty-three of 40 (82.5%) (likely) pathogenic variants were de novo, two were inherited from an affected mother (5%) and five showed unknown inheritance, because parents were not tested (12.5%).

Genes involved in the mutation-positive samples (n=40 (%)).
Cohort 1
For cohort 1, in which 1–5 genes were analysed, 15 (likely) pathogenic variants were found in 231 samples (6.5%). Thirteen (likely) pathogenic variants were found in the PTPN11 gene (5.6%), one pathogenic variant in the RAF1 gene (0.4%) and one likely pathogenic variant in the MAP2K1 gene (0.4%) (table 1).
Mutation-positive samples
Cohort 2
For cohort 2, in which a NGS panel of 14 genes was analysed, 25 (likely) pathogenic variants were found in 193 samples (13%). Of the 25 (likely) pathogenic variants, 14 were found in the PTPN11 gene (7.3%), four were found in the RAF1 gene (2%) and three were found in the RIT1 gene (1.5%). One (likely) pathogenic variant was present in four separate samples in the SOS1, HRAS, BRAF and SHOC2 genes (each gene 0.5%), respectively (table 1).
Two of the 14 pathogenic variants in the PTPN11 gene were maternally inherited. Both mothers were suspected of having Noonan syndrome when the fetal DNA sample was sent in for rasopathy testing.
Distribution of NT between negative and positive samples in cohort 2
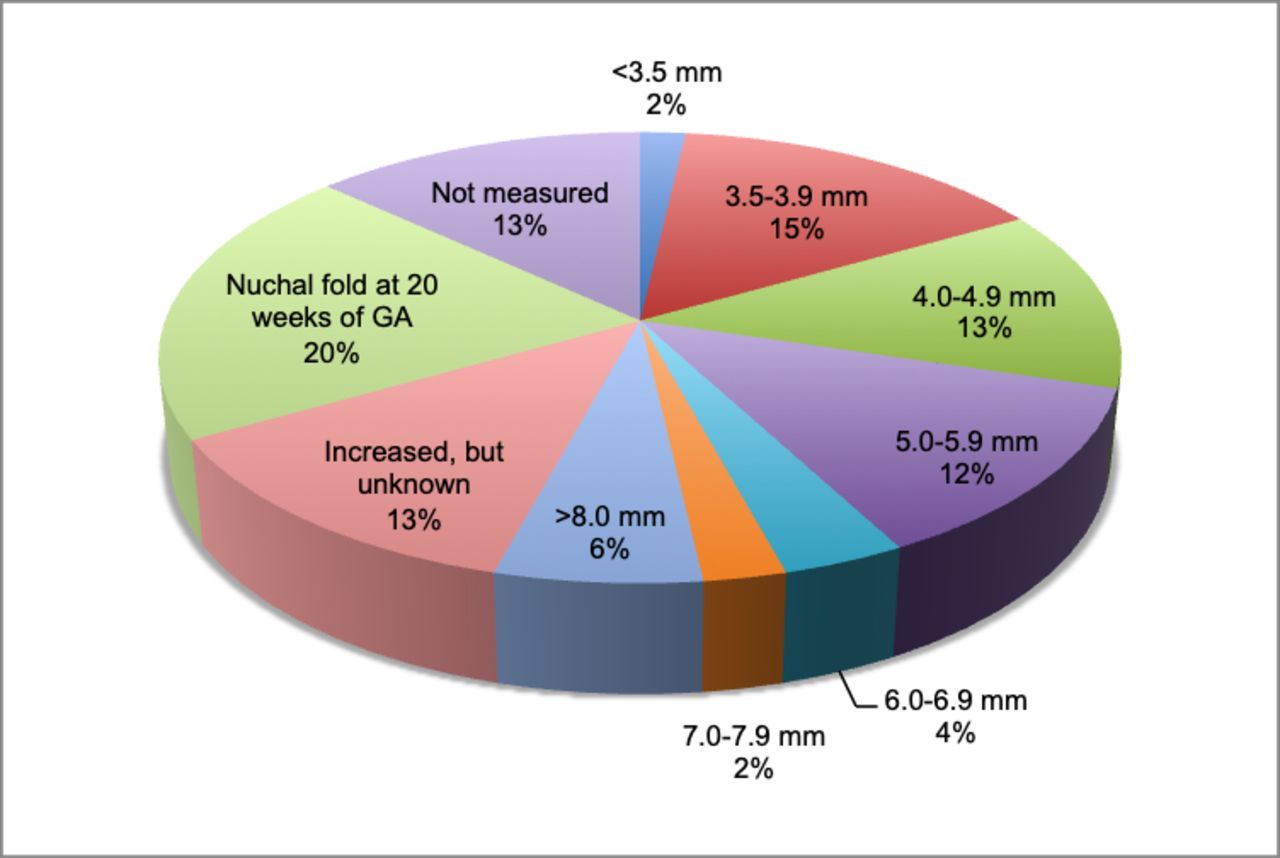
The distributions of NT in cohort 2 were evaluated comparing the mutation-negative and mutation-positive samples (table 2, figures 2 and 3). Tests for normality on samples with measured NT (n=110) showed that mutation-positive samples were normally distributed (mean of 8.46 mm with ±3.7 SD), while mutation-negative samples were non-normal (median of 4.7 mm with ±2.0 IQR) where ~50% of mutation-negative samples had an NT <5 mm. As a result, NT thickness in mutation-positive samples was significantly larger compared with mutation-negative samples (p<0.001, Z=3.858).
Distribution of NT in cohort 2 (mutation-negative versus mutation-positive samples)

Distribution of nuchal translucency in cohort 2: mutation-negative samples (n=168 (%)).

{kind=link}
{kind=link}
{kind=link}
Distribution of nuchal translucency in cohort 2: mutation-positive samples (n=25 (%)).
Clinical findings of all samples in cohort 2
The ultrasound findings of all samples in cohort 2 were analysed according to mutation-positive and mutation-negative samples (table 3). Increased NT/cystic hygroma in the first trimester was the most prevalent finding in all samples (86% of all samples). Although there is a statistically significant difference in the thickness of the NT between the mutation-positive and mutation-negative group, there was no statistic difference in the presence of an increased NT/cystic hygroma between mutation-positive and mutation-negative samples. The ultrasound finding ascites was not statistically more prevalent in the mutation-positive samples. All other ultrasound findings (JLS, hydrops fetalis, pleural effusion, cardiac anomalies, renal abnormalities and polyhydramnios) were statistically more prevalent in the mutation-positive samples (table 3).
Clinical findings in mutation-positive fetuses in cohorts 1 and 2
The 40 pathogenic mutation positive fetuses showed a variable degree of involvement of prenatal findings. Increased NT/cystic hygroma (including persistent NT (defined as NT still visible on ultrasound at 14 weeks of gestation)) was the sole finding in eight fetuses (20.0%). The NTs were between 5.5 and 13 mm for seven samples, whereas one showed oedema around the fetus without exact measurement. Five of these fetuses were terminated at 16 and 22 weeks of gestation, respectively, without autopsy, therefore eliminating follow-up, and one resulted in a premature delivery at 17 weeks of gestation. Two fetuses were life born. In this isolated increased NT group, seven fetuses harboured a pathogenic PTPN11 variant and one a pathogenic RIT1 variant. Nine fetuses were shown to have an increased NT/cystic hygroma with one other finding (22.5%): three had JLS, two had hydrops, one had polyhydramnios, one had a cardiac defect, one showed bilateral hydrothorax and one had unilateral clubfoot. Seven fetuses harboured a pathogenic PTPN11 variant and two a pathogenic RAF1 variant. The remaining 23 fetuses had at least two or more ultrasound findings (23 out of 40 fetuses, 57.5%) (table 4). Although ductus venosus anomalies have been reported as being associated with Noonan syndrome, we found evidence in only one of the mutation-positive fetuses in our study.20–22 This fetus, however, also showed 6 of the specific rasopathy ultrasound features.
All 40 mutation-positive samples were analysed for clinical outcome (table 4). Twenty-eight pregnancies were terminated before 24 weeks of gestation (70%). Two fetuses were born prematurely and died shortly after birth (at 17 and 27 weeks of gestation, respectively)(5%), two pregnancies ended in intrauterine fetal death (5%) and eight pregnancies ended in a term birth (20%). Of these births, one child died during birth, one shortly after birth and one child a few years after birth. Five children with prenatally confirmed Noonan syndrome are still alive (12.5%). There were no significant differences in clinical findings in mutation positive samples cohort 1 versus cohort 2. It was expected that cohort 2 included fetuses with more ultrasound findings due to proposed new criteria by Croonen et al.10 However, not all genetic centres in the Netherlands used these new criteria. Additionally, we did not find a correlation between genotype and phenotype. There were a few recurrent mutations and within those mutations a shared phenotype did not appear to be present. Clinical information about the non-pathogenic variants is provided in the online supplementary table 1.
Supplemental material
Mutation-positive samples (cohorts 1 and 2) and the detected ultrasound findings
Discussion
Recently, a new Dutch guideline was implemented with regard to prenatal testing for a rasopathy. This guideline showed that only a handful of studies were informative for rasopathy testing and it therefore was the motivation for this study. We present the largest cohort of prenatally tested samples for rasopathies. In a previous paper by Croonen et al, testing for a rasopathy in pregnancy was recommended when an enlarged NT is present and at least one of the following features: distended JLS, cystic hygroma, hydrops fetalis, hydrothorax, cardiac anomalies, renal anomalies, polyhydramnios and ascites.10 In this paper, we provide updated recommendations for testing for a rasopathy based on our clinical and genetic findings in a cohort of 424 fetuses.
Mutation detection
In our cohort of 424 fetuses, an overall pathogenic mutation detection rate of 9.4% was found, with a 6.5% mutation detection rate in cohort 1 (testing of 1–5 genes) and 13% in cohort 2 (NGS panel cohort). Croonen et al found a 17.3% mutation rate in their diagnostic group tested for Noonan syndrome. It is unclear what causes the difference in mutation detection rate. The inclusion criteria might play a role, because no isolated enlarged NT was included in their study. However, Croonen et al included cystic hygroma as a separate entity, whereas in our study, these anomalies were combined. If all 58 isolated increased NT samples with NT <5.0 mm had been excluded in our cohort 2, mutation detection in this cohort would have increased to 18.5% (25/135). Additionally, in cohort 1, 1–5 genes were tested, whereas in cohort 2, 14 genes were tested. This latter presumably leads to a higher mutation detection rate. If in cohort 2 only the five genes from cohort 1 were tested, the percentage of mutation detection in cohort 2 would have been 9.8% compared with 13% with NGS panel. However, not every sample in cohort 1 was tested for five genes. Therefore, the mutation detection percentage in cohort 1 might be an under-representation compared with the same genes tested in cohort 2. The difference in mutation detection between our two cohorts seems to be due to the increased amount of genes tested.
In addition to the Croonen et al’s paper, Ali et al recently published a cohort of 39 fetuses with enlarged NT and normal karyotype which underwent testing for Noonan syndrome.16 The authors used two laboratories with a selected panel of 11 and 9 genes, respectively. They found a mutation percentage of approximately 10%. This is the approximately same percentage as we found in our much larger total cohort. However, in our cohort 2 we found a slightly higher mutation detection of 13%. This might be due to the fact that the RIT1 gene was not present in one of the panels used by Ali et al. The RIT1 gene is an important gene in our cohort, with three of the 25 mutations (12%) found in cohort 2 being in this gene. Additionally, microdeletions/duplications were not tested by Ali et al, which have been filtered out in our cohorts.
More than 80% of our mutation-positive samples were de novo. In literature, the de novo rate has been estimated at 30%–75%.6 23 24 Our cases were prenatally detected and the higher percentage could be explained by the fact that milder cases might not show a prenatal phenotype and therefore are not tested in pregnancy. As well, hydropic fetuses have not always been tested for a rasopathy, which would then be missed out on de novo mutation detection.
Genes
In cohorts 1 and 2 combined only eight genes were involved in prenatally confirmed Noonan syndrome. Although no pathogenic variants were found in the A2ML1, CBL, MAP2K1, NRAS, SPRED1 and KRAS genes in our total cohort, in literature a prenatal phenotype has been described for all these genes, except for the SPRED1 gene. For the A2ML1 gene, however, only one report has been published and it is debatable whether this gene is involved in the rasopathies.10 15 25–27
Overall, the PTPN11 gene is prenatally and postnatally the most prevalent gene in the panel; 67.5% of our samples with a rasopathy have a pathogenic PTPN11 variant, which is in agreement with literature.5 6 28
In our study, RAF1 and RIT1 are the most frequently described genes involved in rasopathies after the PTPN11 gene. RIT1 has relatively recently been discovered and it also has an important role in prenatal rasopathies. This is possibly due to the presence of lymphatic malformations in this gene,29 which is easily recognisable on fetal ultrasound. In our study, all three fetuses with a pathogenic RIT1 mutation showed lymphatic problems. However, the indication of sending in samples for testing was increased NT or associated lymphatic problem; therefore, there is a bias in our cohort regarding this ultrasound feature. Only one of the three fetuses with a pathogenic RIT1 variant in our cohort showed a cardiac defect. This is not significantly more than the fetuses with other pathogenic variants. Cardiac anomalies are frequently documented in patients with a pathogenic RIT1 mutation.29 30 However, pulmonic valve stenosis and hypertrophic cardiomyopathy, the common cardiac defects described in Noonan syndrome, are difficult to visualise on fetal ultrasound. Therefore, they are easily overlooked in pregnancy and might not be confined to only fetuses with a RIT1 mutation. SOS1 has to our knowledge only been described a handful of times before in the prenatal setting,15 and in our study only one fetus showed a pathogenic variant. This is possibly due to a milder phenotype, which might not be recognisable on prenatal ultrasound.
Clinical differences between the mutation-negative and mutation-positive group
The prenatal features persistent increased NT, JLS, hydrops fetalis, pleural effusion, cardiac anomalies and polyhydramnios are significantly more present in fetuses with confirmed rasopathy versus fetuses without a confirmed diagnosis. In both the mutation-positive and mutation-negative groups, an increased NT was the most frequent finding on ultrasound, and there was no significant difference between the groups (p-value 0.282). This is due to the fact that in most instances the indication for prenatal testing of a rasopathy was an increased NT. Therefore, it is difficult to confirm if most prenatal rasopathy cases have indeed an enlarged NT. Ascites is not very specific for fetuses with a rasopathy with a p-value of 0.069. Renal anomalies are significantly more present in mutation-positive samples, but it is not a strong predictor (p-value 0.027). Both ascites and renal anomalies are always seen in combination with hydrops fetalis in our mutation-positive and mutation-negative cohort. Additionally, ascites is only detected in a small amount of mutation-positive and mutation-negative fetuses. The most common renal anomaly is pyelectasis. Although pleural effusion is significantly more present in fetuses with a confirmed rasopathy, this feature on its own does not predict a mutation. We did not find mutations in fetus with solely pleural effusion. Pleural effusion is almost always part of hydrops fetalis (tables 3 and 4).
Cohort 2: prenatal ultrasound findings in fetuses with normal chromosomal microarray and analysed for rasopathy using next generation sequencing panel
Clinical findings in the mutation-positive group
In the mutation-positive group, there were eight fetuses (20.0%) with just an isolated NT and only two of these fetuses turned into a term life birth. The other fetuses were terminated before 22 weeks of gestation. It is therefore difficult to determine whether these fetuses would have developed more anomalies in due course of the pregnancy. Twenty-three fetuses showed two or more ultrasound findings additional to the increased NT. Thus, the majority of samples with a confirmed rasopathy (23 of 40 fetuses, 57.5%) have multiple anomalies on ultrasound. We therefore recommend testing for a prenatal rasopathy when the NT is ≥3.5 mm and one of the following ultrasound anomalies is present: JLS, hydrops fetalis, pleural effusion, cardiac anomalies, polyhydramnios. It is debatable whether to test for a rasopathy when an enlarged NT is seen in combination with solely ascites or renal anomalies. The chance of finding a pathogenic variant is low, but not excluded.
A rasopathy cannot be excluded when only one ultrasound anomaly is seen, as our study shows pathogenic variants in eight fetuses with an isolated increased NT (with or without persistent NT enlargement). This has been described before.15 The NTs of the fetuses were, however, large (5.5–13.0 mm). There is a statistical significant difference of the thickness of the NT between the mutation-positive versus the mutation-negative samples with a cut-off at 5.0 mm. Therefore, we recommend testing for prenatal rasopathy in fetuses with an isolated increased NT of ≥5.0 mm. Additionally, in the presence of other ultrasound anomalies, the NT tends to be larger as well (online supplementary file 1), which has been described before.16 Sixteen of the 40 fetuses (40%) with confirmed rasopathy had a cardiac defect. In literature, postnatally, approximately 80% of patients with established rasopathy show a cardiac anomaly.31 The difference is most likely due to the fact that the two most common heart defects in a rasopathy, pulmonic valve stenosis and hypertrophic cardiomyopathy, are difficult to detect on prenatal ultrasound. One fetus with a confirmed pathogenic HRAS variant had a NT of 3.4 mm. This is technically not an increased NT. However, this fetus showed multiple other ultrasound anomalies, fitting the prenatal phenotype of a rasopathy. Prenatal testing for a rasopathy when an increased NT is absent should be based on the remaining ultrasound anomalies. One can consider testing for rasopathies when multiple anomalies described in this study are present.
A genotype–phenotype correlation could not be established in our cohort. This might be due to the fact that many pregnancies, some at early gestation, were terminated when a mutation was found and therefore a complete prenatal and postnatal phenotype is lacking. The high percentage of pregnancy terminations (80%) in the mutation-positive group can have several reasons. An important reason might be that parents, who, due to cultural or religious beliefs, would not terminate a pregnancy in the first place, might not opt for genetic testing. Therefore, this could cause a bias in the percentage in termination of pregnancy. Another reason might be poor prognosis and parental anxiety either due to the enlarged NT or due to the combination of several ultrasound findings. For this reason, however, one would expect also a high percentage in termination of pregnancy irrespective of the presence of a mutation in a rasopathy gene. Unfortunately, we did not follow-up on the mutation-negative samples in our cohort. All parents were counselled by clinical geneticists, and in our opinion, a clinical geneticist is the expert physician for counselling a prenatal rasopathy. Parents will then be able to make a well-informed decision.
Several studies have documented a range of 21%–50% prenatal phenotype in patients with a postnatally confirmed rasopathy. These studies also showed a lack of an association between prenatal severity and postnatal outcome.12 32
Challenges
The requisition forms for cohort 1 were not digitally available. As a result, we did not research the clinical phenotype of the mutation-negative samples in this cohort. In cohort 2, the requisition forms were digitally available and we based the prenatal phenotype on what the referring physician had written down. We trusted the information on the requisition form for the mutation-negative samples to be complete.
Our study did not include newborns with Noonan syndrome or an associated disorder. Additionally, a high percentage of terminations of pregnancy in our cohorts was established, which made postnatal follow-up impossible. Therefore, a complete genotype–phenotype correlation can not be made. Additional research about the postnatal phenotype combined with the prenatal phenotype needs to be performed. Finally, the upcoming use of non-invasive prenatal testing (NIPT) presumably decreases the request for prenatal rasopathy testing as increased NT is an important ultrasound finding in the prenatal rasopathies, which is not measured anymore when performing NIPT.
Recommendations
This study is the most extensive study to date involving the largest cohort of prenatally tested rasopathy. We conclude that all but one confirmed prenatal cases of rasopathy show an enlarged NT. We recommend testing of fetuses with solely an increased NT after chromosomal abnormalities have been excluded when the NT is ≥5.0 mm. We also recommend testing when the NT is ≥3.5 mm and at least one of the following anomalies is present: distended JLS, hydrops fetalis, polyhydramnios, pleural effusion and cardiac defects. Ascites and to a lesser degree renal anomalies in combination with an increased NT are poor predictors to find a pathogenic variant and testing is then debatable. Research for better correlation between prenatal and postnatal phenotype should be performed.
In general, an NGS panel of known rasopathy genes should be used when a rasopathy is suspected. Although we did not find pathogenic variants in every gene in the panel, in all genes, a prenatal phenotype has been documented in literature. Therefore, a smaller panel is not advisable. However, in countries where an extensive panel is not available, testing for only PTPN11 gene would catch of at least 50% of the fetuses with a rasopathy.
We assume that adding recently discovered rasopathy genes (PPP1CB, SOS2 and LZTR1 genes) to the panel would increase the detection rate.
Acknowledgments
The authors would like to thank all referring physicians for their detailed information: Katelijne Bouman, Merel van Maarle, Klaske Lichtenbelt, Karin Diderich, Nicolette den Hollander, Laura van Loon, Gretel Oudesluijs, Anja Kattentidt, Wendy van Zelst-Stams, Marjolein Willemsen, Yvonne Arens, Lutgarde Govaerts, Alexa Vermeer, Chantal Kerkhofs, Floor Duijkers, Emilia Bijlsma, Sonja de Munnik.
References
Footnotes
Contributors Conceptualization: KES and TR. Formal analysis: KES. Investigation: KES and TR. Writing - original draft preparation: KES. Writing - review and editing: KES, TR, MJ, HME, MWE, IvdB, HY.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.