Article Text

Abstract
Background Meiotic homologous recombination (HR) plays an essential role in gametogenesis. In most eukaryotes, meiotic HR is mediated by two recombinase systems: ubiquitous RAD51 and meiosis-specific DMC1. In the RAD51-mediated HR system, RAD51 and five RAD51 paralogues are essential for normal RAD51 function, but the role of RAD51 in human meiosis is unclear. The knockout of Rad51 or any Rad51 paralogue in mice exhibits embryonic lethality. We investigated a family with meiotic arrest, azoospermia and infertility but without other abnormalities.
Methods Homozygosity mapping and whole-exome sequencing were performed in a consanguineous family. An animal model carrying a related mutation was created by using a CRISPR/Cas9 system.
Results We identified a 1 bp homozygous substitution (c.41T>C/p.Leu14Pro) on a RAD51 paralogue, namely, XRCC2, in the consanguineous family. We did not detect any XRCC2 recessive mutation in a cohort of 127 males with non-obstructive-azoospermia. Knockin mice with Xrcc2-c.T41C/p.Leu14Pro mutation were generated successfully by the CRISPR/Cas9 method. The homozygotes survived and exhibited meiotic arrest, azoospermia, premature ovarian failure and infertility.
Conclusion A XRCC2 recessive mutation causing meiotic arrest and infertility in humans was duplicated with knockin mice. Our results revealed a new Mendelian hereditary entity and provided an experimental model of RAD51-HR gene defect in mammalian meiosis.
- RAD51
- XRCC2 mutation
- meiotic arrest
- infertility
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Infertility affects approximately 7% of men worldwide,1 and nearly 50% of infertility cases are associated with genetic defects.2–4 Azoospermia is the main cause of male infertility. To date, mutations on several genes, including SYCP3,5 NR5A1,6 TDRD9,7 TAF4B and ZMYND15,8 TEX11,9 10 DMC1,11 TDRD7,12 MAGEB4,13 SYCE1,14 SOHLH1 15 and MCM8,16 have been identified as the causes of azoospermia. However, the genetic basis of this defect in most infertility cases remains unknown.2–4 12 Meiotic homologous recombination (HR) plays an essential role in proper chromosomal segregation in gametogenesis and enables populations to adapt during evolution by generating new combinations of DNA molecules. Two RecA-related recombinases, namely, DMC1 and RAD51, participate in meiotic HR. DMC1 is a meiosis-specific recombinase,17 whereas RAD51 is involved in mitotic and meiotic HR.18 19 Five RAD51 paralogues, namely, RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3, have been reported to play indispensable roles in RAD51-mediated HR,20 but the detailed mechanism of RAD51-HR in meiosis remains unclear. Studies on RAD51-HR in meiosis are limited because the mammalian knockout of any RAD51 or RAD51 paralogues is embryonically lethal.21–25 In the present paper, we described a RAD51 paralogue point mutation that causes meiotic defects, that is, a XRCC2 recessive mutation (c.41T>C/p.Leu14Pro) leading to meiotic arrest, azoospermia and infertility in humans and mice. Given that humans and mice with XRCC2 mutation survive and show no phenotypes other than reproductive defects, we believe that c.41T>C substitution affects the meiosis-specific function of the XRCC2 protein.
Materials and methods
Subjects and clinical assessments
This study was approved by the ethics committee of the Hunan Children’s Hospital. Before the study, all participated subjects signed a written informed consent. This study included an infertile family (details described in RESULTS section) and 127 sporadic patients (five from Chinese Tujia Miao ethnicity and 122 are from Chinese Han-ethnicity) with non-obstructive azoospermia. These 127 patients were prospectively recruited at Jiahui Genetics Hospital, State Key Laboratory of Medical Genetics, Central South University, Changsha City, China (JGH-SKLMG) from January 2008 to October 2016. At the JGH-SKLMG, after all the azoospermic cases were clinically examined for the anatomical integrity of genital system, the patients who had any history of childhood disease, environmental or radiation exposure, prescription drug usage that could account for their infertility, and other pathologies as varicocele or cryptorchidism were excluded from the investigation. The patients were diagnosed with the clinical findings and several parameters: (1) the conducting of two separate semen analyses (3 weeks apart, each following 3 days of sexual abstinence, according to the WHO criteria, WHO laboratory manual for the examination of human semen and semen-cervical interaction 1999), (2) the percutaneous epididymal sperm aspiration, (3) the blood hormone analysis that included follicle stimulating hormone, luteinising hormone, prolactin and testosterone, (4) the karyotype analysis that was performed with G-banding methods at 550 bands level, for each karyotype analysis, at least 30 peripheral blood metaphases were analysed, (5) the microdeletions in the AZF region and the SRY gene region of the Y chromosome were screened by regular PCR.
DNA isolation
Peripheral-blood samples for DNA extraction were obtained from 18 family members as described in figure 1A and 27 sporadic cases of non-obstructive azoospermia according to a standard protocol.
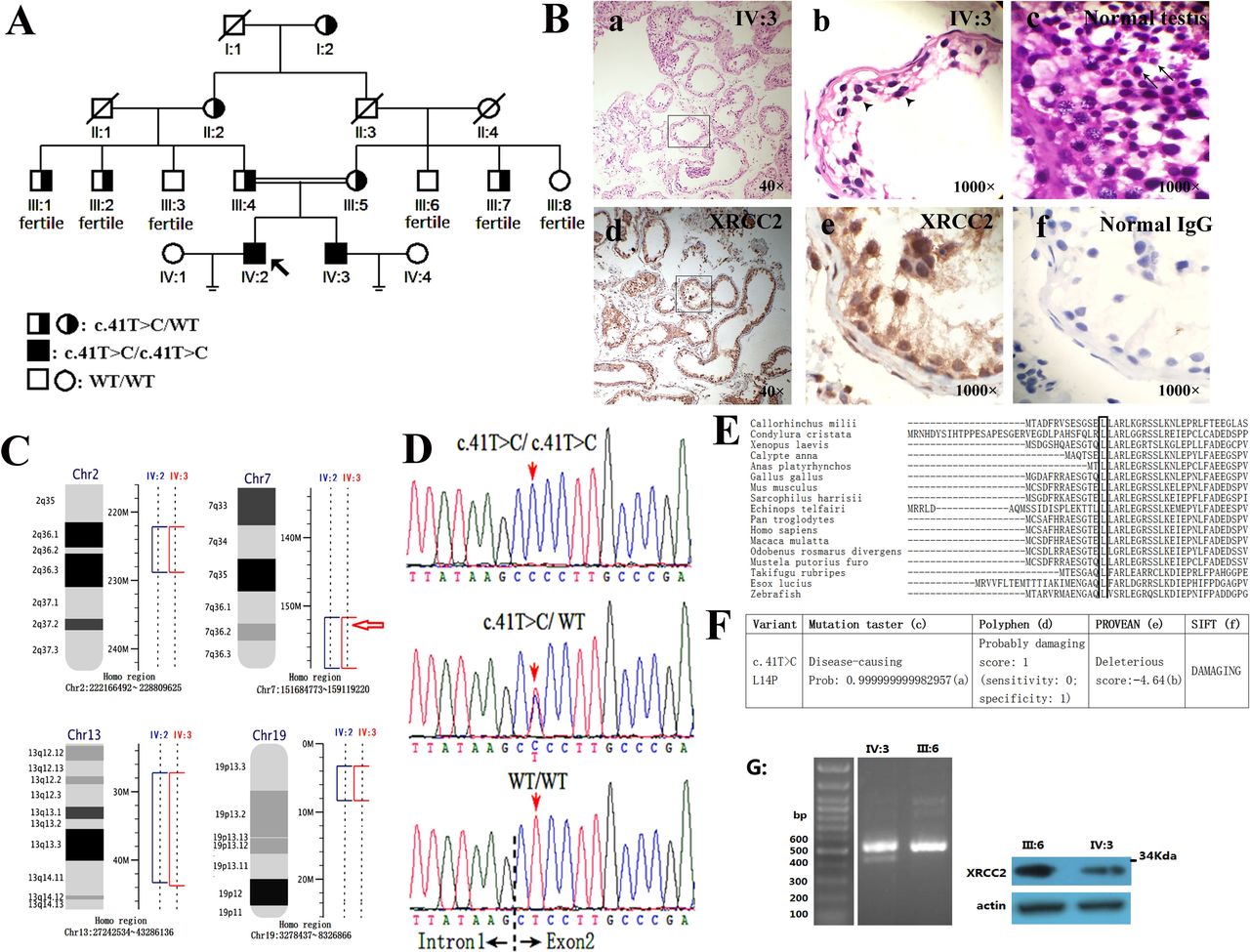
Mutated XRCC2 in a family with meiotic arrest, azoospermia and infertility. (A) Pedigree with XRCC2 mutation. (B) Histology of human seminiferous tubules. Magnification: a,d=40×; b,c,e,f=1000×. (a,b) H&E staining disclosed that the tubules of IV:3 (29 years old) were irregularly shaped. No mature sperm was observable, and the cells were arrested at the zygotene stage of meiosis prophase I (arrowhead). (c) H&E staining of tubules in normal human testis (67 years old). The cells at least proliferated to the stage of round spermid (arrow). (d,e,f) Immunohistochemistry of XRCC2 in tubules of IV:3. Sections immunohistochemically labelled for XRCC2 (in brown) (d,e) or an IgG antibody as negative control (f). (C) Linkage mapping in the family disclosed the homozygous intervals shared by two patients (IV:2 and IV:3). Red arrow: Human XRCC2 position. (D) Chromatograms of XRCC2 mutations by Sanger sequencing. (E) Evolutionary conservation of the p.Leu14 residues of XRCC2 protein. (F) Functional predictions of XRCC2-p.Leu14Pro substitution: (a) Probability value mean probability of prediction, the value close to 1 indicates a high security of the prediction. (b) PREDICTION (cut-of =−2.5). (c) http://www.mutationtaster.org/. (d) http://genetics.bwh.harvard.edu/pph2/. (e) http://provean.jcvi.org/genome_submit_2.php. (f) http://sift.jcvi.org/www/SIFT_chr_coords_submit.html. (G) RNA and protein assay on peripheral blood lymphocyte. L: Fragments amplified from the reverse transcribed cDNA sample of IV:3 and III:6 and then transferred on 0.8% agarose gel. Note: Two bands were observed in IV:3. R: Western blot detected the full XRCC2 protein on IV:3 and III:6.
Homozygosity mapping
Whole-genome genotyping of six family members (I:2, II:2, III:4, III:5, IV:2 and IV:3 in figure 1A) was carried out to identify the presence of runs of homozygosity (ROH). Briefly, 250 ng of genomic DNA per sample was processed on an Illumina Human Omni ZhongHua-8 Beadchip array (Illumina, San Diego, California, USA) according to a standard protocol. Normalised output was generated using Genome Studio V.2011.1 (Illumina). The plink software (http://zzz.bwh.harvard.edu/plink/ibdibs.shtml#homo) was used to search for ROH regions according to established criteria.
Whole-exome sequencing
Whole-exome sequencing (WES) was carried out for four family members (III:4, III:5, IV:2 and IV:3, figure 1A). In brief, genomic DNA (approximately 1.5 ug) for each member was fragmented to an average size of 350 bp and subjected to DNA library creation using established Illumina paired-end protocols. Adaptor-ligated libraries were amplified and indexed via PCR. Then the library was hybridised to NimbleGen SeqCap EZ library Exome Capture Kit (44M, Roche, Madison, USA), and paired-end 90-base massively parallel sequencing was carried out on an Illumina HiSeq 2000, according to the manufacturer’s protocols. The reads mapping and variants analysis pipeline is as follows. Raw reads with low quality or containing adapters were filtered before mapping. For SNP calling, filtered reads were aligned to the human genome reference (UCSC hg19) with Short Oligonucleotide Analysis Package (SOAP, V.2.21)26 and then SOAPsnp software (V.1.05)27 was used to detect SNPs. We eliminated low quality SNPs with a score of genotype quality less than 20 or the sequencing depth is smaller than 4. For indel calling, Burrows–Wheeler Aligner28 was used to do the alignment. Duplicate reads were marked using the Picard toolkit and Genome Analysis Tool Kit (GATK)29 was used to call small indels.
Sanger sequencing
Sanger sequencing was performed on an ABI-A3500 genetic analyzer, and a BigDye 3.1 sequencing kit (Applied Biosystems, California, USA) was used. The primers used for PCR and Sanger sequencing are provided in online supplementary table S1.
Supplemental material
Generation of Xrcc2-c.41T>C/p.Leu14Pro mice
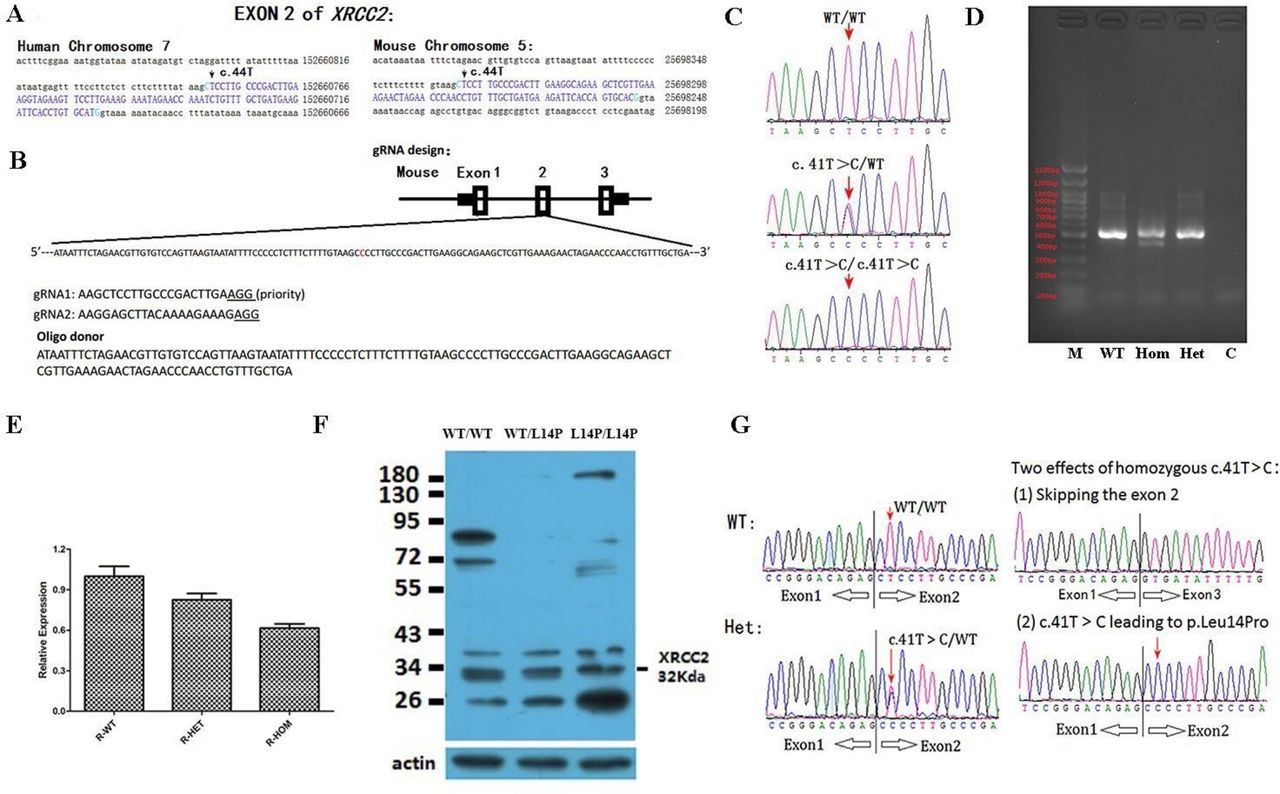
C57BL/6 mouse model with point mutation (c. 41 T>C) at Xrcc2 locus was generated by microinjection of CRISPR/Cas9 into fertilised eggs (Cyagen, China). The mouse Xrcc2 gene (GenBank accession number: NM_020570.2; Ensembl: ENSMUSG00000028933) was located on chromosome 5qB1. The base c.T41 mutation in oligo donor was introduced into exon 2 by homology-directed repair. Cas9 mRNA, gRNA and oligo donor were coinjected into the zygotes for KI mouse production (figure 2A,B). The pups were genotyped by PCR and Sanger sequencing (primers and conditions, see online supplementary table S1) using the genomic DNA or cDNA as template.

Generation of Xrcc2 knockin mice by CRISPR/Cas9-mediated genome engineering. (A) Genomic sequence characteristics nearing Xrcc2 exon 2. (B) Schematic of target site at mouse Xrcc2 locus. In donor DNA, the single-strand oligo-deoxynucleotide, namely, the replaced nucleotide, was observed. (C) Chromatograms of Xrcc2 mutations from PCR-amplified fragments from mouse tail genomic DNAs. (D) Fragments amplified from the reverse transcribed cDNA sample of mice testes and then immigrated on 0.8% agarose gel. Note: Two bands for homozygotes were observed. (E) Mean expression (±SEM) of Xrcc2 transcripts (n=3) in three groups of pubs (P<0.05). Note: approximately 70% of normal Xrcc2 transcripts were observed on homozygous mutant testes mRNA. (F) Western blot detected the Xrcc2 proteins in all three groups of testes. Identically sized bands are detected in WT, heterozygote and mutant testis lysates, including a band at the size expected for Xrcc2, strongly suggesting the mutant Xrcc2 protein is present in the testes of homozygous mutant mice. (G) Reconfirmation of Xrcc2-c.41T>C in heterozygotes and homozygotes in the RNA level. To amplify the fragment from cDNA, the forward primer is located on exon 1, the reverse primer is located on exon 3, the sequencing primer is the forward primer. Note: For homozygote, two bands were amplified, which were sequenced individually. The results indicated the exon 2 was skipped for some of the Xrcc2 transcripts. Note: For figure CDEFG, the mice age is 90 days. WT, wild type.
To minimise the risk of off-target effects caused by CRISPR-Cas9, we used the following strategies. (1) We used a gRNA with a high score of 85. The higher the score was, the lower the likelihood of the off-target effects would be. (2) We generated four F0 mice, namely, F2, M3, M21 and F22. Of these four F0 mice, M3 and M21 males were sterile. The two remaining F0 females (F2 and F22) were subsequently used as founders of two independent strains and backcrossed with wild type (WT) mice for two generations to produce NF2 descendants that were intercrossed to generate Xrcc2 L14P/L14P, Xrcc2 L14P/WT or Xrcc2 WT/WT mice. (3) We transmitted Xrcc2-c.41T>C mutation in mice for six generations and found that the azoospermia phenotype segregated with the homozygote genotype in the two Xrcc2-KI mouse lines (F2 and F22), respectively.
All the animal experiment protocols of this study were approved by the Animal Experiment Committee of Hunan Children’s Hospital (Changsha, China). All experiments were performed in accordance with the approved guidelines and regulations.
Histology and TUNEL assay
Mice ovaries or testes were fixed in Bouin’s solution. After dehydrated through an ethanol series, tissues were embedded in paraffin and serially sectioned, and stained with H&E. For TUNEL assay, the In Situ Cell Death Detection Kit (Kit for immunohistochemical detection and quantification of apoptosis at single cell level, based on labelling of DNA strand breaks, Cat. 11684817910, Roche Applied Science) was used according to the manufacturer’s instructions.
Immunocytochemistry
Testicular tissue sections from IV:3, which is a patient with infertility belonging to the family (figure 1A), WT mice and Xrcc2 L14/L14P mice were immunereacted with a primary anti-XRCC2 antibody (1:100 dilution of rabbit polyclonal antibody, Gene Tex,GTX16397) to assess the XRCC2 expression in the testes. The test of affinity of the XRCC2 antibody (Gene Tex, GTX16397) was performed by over expression assay (online supplementary figure S1).
The primary antibody was detected using an HRP-labelled goat anti-rabbit IgG polymer (PV-6001, ZSGB-BIO, China). HRP interacting with hydrogen peroxide and dark brown DAB was visualised on a microscope in accordance with the product protocol.
Western blotting
Protein lysates from control and mutant testes (mice) or from peripheral blood lymphocyte (PBL) (human) were prepared. The total protein content was determined with the Bradford method. Each sample was subjected to SDS-PAGE and the membrane was immunoblotted with rabbit XRCC2 antibody (GeneTex, GTX16397) at a 1:1000 dilution. Secondary IgG-HRP antibody (1:2000; Santa Cruz Biotechnology) and an ECL Chemiluminescence kit (GE healthcare) were used for signal visualisation.
RNA extraction and real time PCR
Total RNA was extracted from testes tissues (mice) or from PBL (human) using RNAiso Plus Kit (TaKaRa, Dalian, China). cDNA was synthesised from 1 ug of RNA using the PrimeScript II 1st Strand cDNA Synthesis Kit (TaKaRa, Dalian, China). PCR reactions were performed on a 7500 Real Time PCR System (Applied Biosystems) using NovoStart SYBR qPCR SuperMix (Novoprotein, Shanghai, China). The primers were used in the experiments of qPCR: XRCC2-5’UTR-F and XRCC2-3’UTR-R (online supplementary table S1). Each assay was duplicated four times. The data were analysed by unpaired two-tailed t tests using R software.
Chromosome breakage assay
For assays of chromosome breakage in XRCC2L14P/L14P cells and other cells, the PBLs were treated with mitomycin C (MMC, Catalogue#: 2713–5; CAS Number: 50-07-7; BioVision, 155 s. Milpitas Boulevard, Milpitas, California, USA) or Cisplatin (DDP, Cas No.: 15663-27-1; Houston, Texas, USA). Except for a certain amount of DNA breaking agents (MMC or DDP) or the same volume of PBS added into the RMPI-1640 medium, all the cell-culture and experimental procedures are according to standard Giemsa banding protocol in medical genetic laboratory.
Results
Human clinic data
A Tujia ethnic minority family with male infertility residing in the Xiangxi Tujia–Miao Autonomous Prefecture, a west region of Hunan Province, China, participated in this study (figure 1A). Small testes were detected in the patients (IV:2 and IV:3) at 10 and 12 years as a result of routine regular health check-ups at schools. The sizes of the testes of IV:2 and IV:3 remained small at the ages of 29 and 31 years (about 9–11 mL), respectively. Infertility in IV:2 and IV:3 was diagnosed by two independent andrologists with the following parameters: (1) two separate semen analyses (3 weeks apart, each following 3 days of sexual abstinence) reported no sperm observed (online supplementary table S2), (2) no sperm was observed in two separate occasions of percutaneous epididymal sperm aspiration (data not shown), (3) testicular biopsy reported no sperm observed and indicated meiotic arrest (figure 1B and online supplementary figure S2) and (4) normal results were reported in other clinical tests (online supplementary table S3). We found no other inherited disease in this family, and the oldest family member (I:2) is a 97-year-old woman.
Homozygosity mapping narrowed down to 35.2 Mb intervals
Given the parental consanguinity for the proband in the family, homozygous intervals were identified (online supplementary figure S3). A total of 35.2 Mb genomic intervals (2q26.1-q36.3, 7q36.1-qter, 13q12.1-q14.1 and 19p13.2–19 p13.3) shared by the two infertile brothers were identified (figure 1C). In these intervals, we did not observe any region showing overlap with the previously described loci of azoospermia, meiotic arrest and infertility. This result implies that one of them may represent a previously undescribed locus of meiotic arrest and infertility.
Whole-exome sequencing (WES) revealed XRCC2 recessive mutation
WES was performed and an average of 4.97 Gb data was yielded within the target region of each sample. The quality statistics for WES are shown in online supplementary table S4. We found 29 241 variants in III:4, 29 350 variants in III:5, 29 361 variants in IV:2, 29 920 variants in IV:3. After merging the shared variants, a total of 40 728 variants were obtained in this family. First, only variants with allele frequency less than 1% in population were considered for subsequent analysis. After screening and filtering the following databases, that is, gnomAD_exome_ALL, gnomAD_exome_EAS, gnomAD_genome_ALL, gnomAD_genome_EAS, Freq_ExAC_ALL, Freq_esp6500siv2_all and Freq_1000g2015aug_all, only 2441 variants were left. We then limit the variants in coding/splicing regions (removing intergenic, intronic, upstream and downstream variants), the number of variants was reduced to 1968. After removing benign or likely benign variants predicted by InterVar,30 1444 variants were left. Among the left variants, only 10 homozygous variants were shared by the two azoospermic men (online supplementary table S5). Further excluding the variants that are homozygous in either parent, only one variant was left (online supplementary table S6).
The brief filter strategy was provided in online supplementary figure S4.
This variant was a 1 bp substitution on XRCC2 (c.41T>C; Chr7:152357866 T>C). Coincidentally, XRCC2 gene is located in the 7q36.1-qter homozygous interval (figure 1C).
XRCC2 sanger sequencing and functional analysis
Sanger sequencing confirmed the XRCC2-c.41C>T mutation and showed that it cosegregated with azoospermia in the family (figure 1A). In accordance with the reference sequence (NM_005431), the c.41T>C changed codon 14 from CTC to CCC (figure 1D), resulting in the substitution of leucine by proline (p.Leu14Pro).
Multiple alignment analysis results indicated that residue p.Leu14 was highly conserved among different species (figure 1E). Consistently, functional prediction with four programmes in silico implied that XRCC2- p.Leu14Pro was disease-causing or damaging (figure 1F). Meanwhile, we noticed that the c.41T>C substitution neared the splicing site of exon 2 of the XRCC2 gene (figure 1D).
We attempted to verify whether the XRCC2 protein is still present in the patients’ testes. In seminiferous tubules from the IV:3 patient, the XRCC2 protein was detected by histological analysis (figure 1Bd-f). Total RNA and protein were extracted from PBLs. Consistently, we observed that the full XRCC2 transcripts and the full XRCC2 protein can be detected on extracts of PBLs (figure 1G). XRCC2 is an HR and double-strand break (DSB) repair gene.22 24 31 32 Latest research shows atypical Fanconi anaemia and a marked increase in the frequency of chromosomal DSBs in response to crosslinking agents on a child with XRCC2 truncating mutation.31 32 Accordingly, we quantified the chromosomal breaks induced by two chromosome crosslinking agents, namely, MMC or cisplatin (DPP), on cells from IV:2 and IV:3. However, in lymphocytes cultured with MMC or DPP, the XRCC2-p.L14P cells exhibited similar frequencies of chromosomal breaks to those of normal cells (online supplementary table S7 and figure S5). Simultaneously, we did not find other phenotypes in the family.
To confirm that the XRCC2 mutation caused human meiotic arrest and azoospermia, we performed direct sequencing of XRCC2-coding regions on 127 infertile men with non-obstructive azoospermia. However, none of them carried the c.41T>C mutation or other XRCC2- recessive mutation (data not shown). We then proceeded with the generation of the animal model.
Generation of Xrcc2-c.41T>C/p.Leu14Pro mice
We observed high similarities between human XRCC2 and mice Xrcc2 genomic sequences (figure 2A). The CRISPR/Cas9 method was used to generate knockin mice with the Xrcc2-c.41T>C/p.Leu14Pro mutation (below described Xrcc2 L14P) when the genotypes were Sanger-sequenced at genomic DNA (figure 2C) or mRNA levels (figures 2D, E and G). At the mRNA level, we designed primers that were located on the 5’UTR or 3’UTR region of mice Xrcc2 (online supplementary table S1). When mRNA was extracted from the mice testes, Xrcc2-c.41T>C was reconfirmed in heterozygotes (figures 2D, E and G). In homozygotes, we observed a double effect of Xrcc2: (1) the c.41T>C substitution and (2) skipping of the whole exon 2 (which leading to a truncating protein p.L14Vfs*20) in about 30% of the XRCC2 transcripts (figures 2D, E and G).
Xrcc2 L14P/WT mice intercrosses transmitted the mutation to their offspring at normal Mendelian frequencies (online supplementary table S8, 21 litters, 188 offspring calculated, wt=46, heterozygote=99 and homozygote=43). No difference in weight was found among the three groups of mice at 7, 30 and 60 days postpartum (dpp; online supplementary tables S8–12). Morphologically, the WT, Xrcc2 L14P/WT, and Xrcc2 L14P/L14P animals were indistinguishable at 7, 14, 28 or 35 dpp (representative see online supplementary figure S6).
All of the male Xrcc2 L14P/L14P mice exhibited meiotic arrest, azoospermia and infertility
The male phenotype of Xrcc2 L14P/L14P mice was analysed. We monitored 8-week-old Xrcc2 L14P/L14P males mated with WT mice for 60–90 days and observed that 100% of homozygotes were infertile (online supplementary table S13) (n=17). Afterwards, 28 male homozygotes were further evaluated, and all of the homozygotes were infertile. The infertile Xrcc2 L14P/L14P males (n=10) were sexually active, and this finding was indicated by the repeated detection of vaginal plugs in the mating females. The testes of the Xrcc2 L14P/L14P mice were smaller than those of the WT mice from 21 dpp to 180 dpp (p<0.05). In contrast to WT males weighing 97–104.5 mg (figures 3A and 7), the Xrcc2 L14P/L14P mice weighed 27.3–37.9 mg at 90 dpp. The histological examination of the testes from 90dpp Xrcc2 L14P/L14P males revealed the following results: (1) nearly all of the seminiferous tubules were depleted (figure 3Bc-f); (2) no mature sperm was observed in all of the tubules (figure 3Bc-f); (3) in each testis, the tubules showed multiple layers of spermatocytes arrested in the zygotene and pachytene stages of meiotic prophase I (figure 3Bef and S6ef at 28 or 180 dpp mice) and (4) the TUNEL assay indicated that the number of apoptotic spermatocytes markedly increased in some tubules (figure 3C). The tubules were histologically examined at 7, 14, 21, 28, 90 and 180 dpp. However, no sperm was detected in seminiferous tubules from the homozygotes from 7 dpp to 180 dpp, indicating a complete meiotic arrest (online supplementary figure S7).
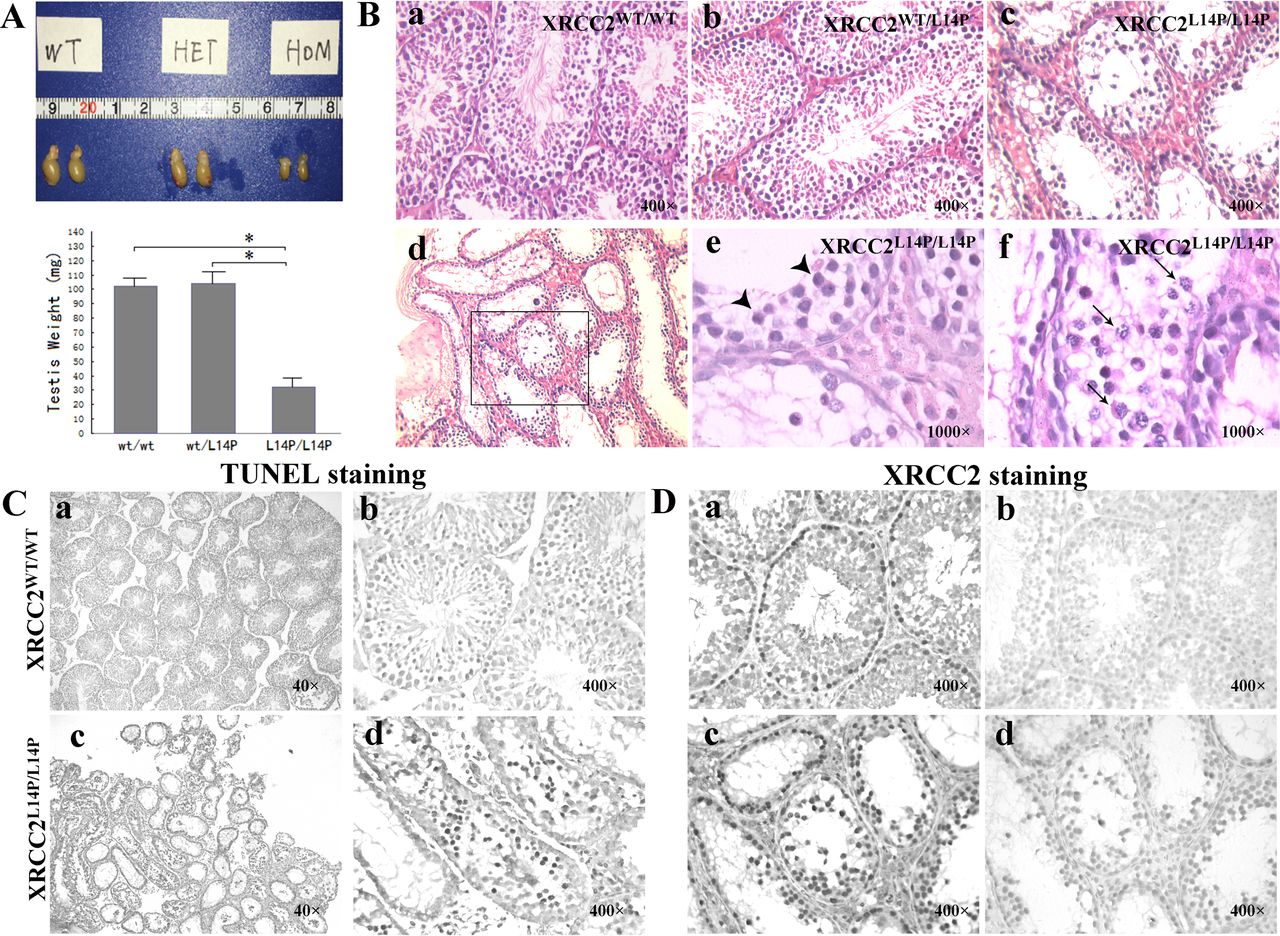
Effects of Xrcc2 L14P in male mice at 90 days. (A) Testis size (up) and weights (down) for respective genotypes are shown. Error bars indicate SD from the mean of the testis weights (n=6). Asterisks represent statistical significance (p<0.05). (B) H&E staining of mice testes. (a) tubules of Xrcc2 WT/WT; (b) tubules of Xrcc2 WT/L14P; (c,d,e,f) tubules of Xrcc2 L14P/L14P, arrowhead: zygotene stage cells; arrow: apoptotic like-pachytene stage cells. Magnification: a,b,c=400×; d=400×; e,f=1000×. (C) TUNEL staining of Xrcc2 WT/WT and Xrcc2 L14P/L14P testes. Apoptotic cells are labelled in dark brown. (a,b) Xrcc2 WT/W testes showed no apoptotic spermatocytes. (c,d) Apoptotic spermatocytes were found in tubules of Xrcc2 L14P/L14P testes. Magnification: a,c=40×; b,d=400×. (D) Testis sections were immunohistochemically labelled for Xrcc2 (ac) (in dark brown) and section stained with IgG antibody (b,d) as a negative control. (a,b) Xrcc2 WT/WT testis; (c,d) Xrcc2 L14P/L14P testis. Magnification: a,b,c,d=400×.
Mutant XRCC2 is present in testes
The WT Xrcc2 protein is highly expressed in testis,33 similar to the above mentioned results at mRNA lever. Consistently, we observed that the Xrcc2 protein was expressed in the inner layer of spermatocytes of mouse tubules (figure 3Da and 3Db) and mutant XRCC2 was detected in Xrcc2 L14P/L14P mouse tubules (figure 3Dc and 3Dd). This observation was consistent with human results, which demonstrated that the mutant Xrcc2 could be visualised in mutant tubules (figure 1Bd-f). In Western blot analysis, the full Xrcc2 protein was present in the testis extracts from homozygotes and other animals (figure 2F).
Half of theXrcc2L14P/L14P females exhibited infertility, whereas the other half showed a low reproductive capacity
We monitored 8-week-old Xrcc2 L14P/L14P females mated with WT mice for approximately 60 days and observed that 47% of the female homozygotes were infertile (online supplementary table S14). The repeated detection of vaginal plugs indicated that the infertile Xrcc2 L14P/L14P females (n=8) were sexually active. However, both ovaries (n=5) were small, atrophic and free of identifiable follicles and fibrosis at 90 dpp for 47% of the infertile females (figure 4B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histology of ovaries showing ovulation defect in Xrcc2 L14P/L14P females. (A) Normal ovary of a littermate control. (B) Ovary of an infertile Xrcc2 L14P/L14P female at 90 dpp. (C,D) Ovary of an fertile Xrcc2 L14P/L14P female at 93 dpp. Note: No corpora lutea or follicle was identifiable in one side of the ovary (C), and few corpora lutea or follicles were observed for the other side of the ovary (D). Magnification: A,B,C,D=40×. Box: ovaries. dpp, days postpartum.
For the 53% fertile Xrcc2 L14P/L14P females, the litter size was significantly reduced, and an average of 4.5 mice was produced by each litter (p<0.01). The mating time to give birth was evidently prolonged to an average of 44 days (p<0.01; online supplementary table S15). Their ovarian abnormalities, including small sizes, atrophy and no identifiable follicles, were prevalent on one side (figure 4C and D).
We further evaluated 60 ovaries from 7 to 180 dpp Xrcc2 L14P/L14P females (online supplementary table S16 and figure S7). The results showed that follicles could be observed in 100% of 7 or 14 dpp ovaries, 80% of 21 dpp ovaries, 70% of 28 dpp ovaries, 50% of 90 dpp ovaries and 20% of 180 dpp ovaries (online supplementary table S16 and figure S8). These data indicated premature ovarian failure.
Discussion
Meiotic HR participates in gametogenesis. In most eukaryotes, meiotic HR is mediated by two recombinase systems, namely, ubiquitous RAD51 and meiosis-specific DMC1. Meiotic HR is initiated by the formation of DNA DSBs that are catalysed by the enzyme SPO11.34–36 DNA ends are then resected at DSB sites. Two RecA-related recombinases, namely, DMC1 and RAD51, form nucleoprotein filaments on a single-strand DNA flanking these DSB sites. They catalyse strand invasion and strand exchange reaction with a homologous interval on another DNA molecule to ensure successful repair. Five other RAD51 paralogues forming at least two stable complexes, namely, BCDX2: RAD51B-RAD51C-RAD51D-XRCC2 and CX3: RAD51C-XRCC3, have been reported to play important roles in RAD51-mediated HR-DSB repair.8 The BCDX2 complex is responsible for RAD51 recruitment or stabilisation at damage sites,20 37 38 whereas the CX3 complex acts downstream of RAD51 recruitment to damage sites associated with Holliday junction resolvase activity, possibly indicating its function of stabilising gene conversion tracts.20 38 DMC1 is a meiosis-specific recombinase,17 whereas RAD51 is essential for mitotic and meiotic recombination.18 19 This role is consistent with the association of an XRCC2 gene variant or other RAD51 paralogue variants with a high risk of cancer.39 Our study did not find any evidence of cancer in the family despite a careful inquiry possibly because the sample size of the family was limited, or the XRCC2 Ler14Pro mutation did not disrupt the HR function of somatic cells.
However, whether the RAD51-HR system uses the same mechanism in mitotic and meiotic cells remains unclear.19 In humans, researchers have yet to determine why two homologous DNA recombinases, namely, RAD51 and DMC1, are required in mammalian meiosis.40 41 To identify the mechanism of HR, we should create a RAD51 mutant model that specifically disrupts only one of the RAD51-HR-DSB repair functions, which are mitosis and meiosis HR-DSB repair functions. Unfortunately, knockout rodents of Rad51 or Rad51 paralogues are embryonically lethal,20–24 so mechanistic studies on RAD51-HR-DSB repair in meiosis are limited. Using a separation-of-function mutant form of Rad51, Veronica Cloud et al 19 reported that Rad51 performs a filament-forming function in HR of meiosis, but the function of homologous joint molecules is unnecessary. This observation probably indicates that Rad51 uses different functions between mitotic and meiotic cells. In the present study, we identified XRCC2 recessive mutations in a family with complete meiotic arrest, azoospermia and infertility. The mouse model with the Xrcc2 L14P mutation replicated the phenotypes. Homozygous female mice exhibited reproductive disorders that were consistent with premature ovary failure. Human XRCC2 and mouse Xrcc2 mutants did not exhibit other identifiable phenotypes. Although Xrcc2-c.41T>C showed 30% splicing changes, the full Xrcc2 protein was detected. We then proposed that XRCC2-Leu14Pro was a meiosis-specific mutation, and this finding was further confirmed by the following aspects.
First, unlike an XRCC2-truncated male showing congenital malformations and atypical Fanconi anaemia, patients with XRCC2-c.41T>C/p.Leu14Pro exhibited infertility only. Shamseldin et al 31 and Park et al 32 identified a truncating XRCC2 (homozygous XRCC2-p.Arg215*) on a 2.5-year-old male who displayed microcephaly, absent thumbs, absent first metacarpal and scaphoid bones, absent radius, facial paralysis, ectopic kidney and severe growth deficiency. The somatic cells of this child displayed a marked increase in the frequency of unrepaired DSBs in response to crosslinking agents. Thus, the child was proposed to be affected with atypical Fanconi anaemia.31 32 In the present study, when the infertile brothers (IV:2 and IV:3) with the XRCC2-c.41T>C/p.Leu14Pro homozygous mutation were examined independently by a neurologist, haematologist and orthopaedist, neither exhibited any of the abovementioned malformations (data not shown). In the assay of the somatic cells cultured with DNA breakage agents, the frequencies of the unrepaired chromosomal breaks of XRCC2-p.Leu14Pro lymphocytes did not increase (online supplementary table S7 and figure S5). Thus, the XRCC2-p.Leu14Pro mutation did not disrupt the HR-DSB repair function in somatic cells, or the effect was minimal that it was hardly distinguished.
Second, in contrast to the Xrcc2 −/− mice exhibiting embryonic lethality, reduced size and morphological abnormalities, the Xrcc2 L14P/L14P mice were viable with unreduced size and did not show identifiable malformation other than reproductive defects. These observations verified that Xrcc2 is involved in HR-mediated DSB repair.22 24 31 32 Deans et al 22 constructed an Xrcc2 knockout model, in which exon 3 (86% ofanXrcc2 coding sequence) is deleted. In more than 300 offspring, heterozygote intercrosses fail to produce any Xrcc2 −/− mice, thereby indicating that Xrcc2 −/− mice are embryonically lethal.22 Additionally, Xrcc2 −/− embryos displaying growth retardation and morphological abnormalities are observed.22 In the present study, the Xrcc2 L14P mouse model revealed that the heterozygote intercrosses transmitted the mutation to the offspring at normal Mendelian frequencies (about 1:2:1; supplementary table S8). The Xrcc2 L14P/L14P mice were viable, and their size, body weights and physical characteristics were indistinguishable among the three groups of offspring (online supplementary tables S8–S12 and figure S6).
Third, p.Leu14Pro is located on the linker region of XRCC2. In RAD51 paralogues, we observed that all of the members except XRCC2 are composed of three domains, namely, N-terminal and C-terminal domains connected by a linker region41 (online supplementary figure S9). However, XRCC2 comprises two domains, namely, a C-terminal domain and a linker region, and lacks an N-terminal domain41 (online supplementary figure S9). Previous studies stated that the linker region of RAD51 paralogues is essential for protein–protein interactions,20 42 and two complexes, namely, BCDX2 and CX3, have been suggested.20 37 38 In the present study, p.Leu14Pro mutation occurred on the linker region of XRCC2 (online supplementary figure S9), and this phenomenon is different from the previously reported truncating p.Arg215*mutation that truncated a part of the C-terminal region of XRCC2 (online supplementary figure S8A) on a patient with atypical Fanconi anaemia.30 32
Therefore, this study identified a novel genetic entity of HR-mediated DNA repair gene mutation, that is, the point mutation on the linker region of XRCC2 was a meiosis-specific mutation causing meiotic arrest and infertility. Further functional studies on the mutation in the XRCC2 linker region should be conducted.
Acknowledgments
The authors are grateful to the family members who participated in this study.
References
Footnotes
YY and JG contributed equally.
Contributors YJY and LQW designed the research. YJY, JHG, LD, YMZ, HH, LHT, WJC, JHG, MT, KWW performed the sample collection and the research. YJY, JHG and LD performed the bioinformatics analysis. YJY and LQW analysed the experimental data and wrote the paper.
Funding This work was supported by grants from the National Natural Science Foundation of China (31501017, to YJY), the Key Nature Science Foundation of Hunan Children’s Hospital (2015-0002, to YJY), the National Natural Science Foundation of China (81771599, to LQW), and the Key Laboratory fund of Hunan Province (2018TP1028, to YYJ).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Hunan Children’s Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.