Article Text

Abstract
Background Heterozygous germline loss-of-function mutations in the aryl hydrocarbon receptor-interacting protein gene (AIP) predispose to childhood-onset pituitary tumours. The pathogenicity of missense variants may pose difficulties for genetic counselling and family follow-up.
Objective To develop an in vivo system to test the pathogenicity of human AIP mutations using the fruit fly Drosophila melanogaster.
Methods We generated a null mutant of the Drosophila AIP orthologue, CG1847, a gene located on the Xchromosome, which displayed lethality at larval stage in hemizygous knockout male mutants (CG1847exon1_3 ). We tested human missense variants of ‘unknown significance’, with ‘pathogenic’ variants as positive control.
Results We found that human AIP can functionally substitute for CG1847, as heterologous overexpression of human AIP rescued male CG1847exon1_3 lethality, while a truncated version of AIP did not restore viability. Flies harbouring patient-specific missense AIP variants (p.C238Y, p.I13N, p.W73R and p.G272D) failed to rescue CG1847exon1_3 mutants, while seven variants (p.R16H, p.Q164R, p.E293V, p.A299V, p.R304Q, p.R314W and p.R325Q) showed rescue, supporting a non-pathogenic role for these latter variants corresponding to prevalence and clinical data.
Conclusion Our in vivo model represents a valuable tool to characterise putative disease-causing human AIP variants and assist the genetic counselling and management of families carrying AIP variants.
- AIP
- drosophila melanogaster
- FIPA
- pathogenic genetic variant
- pituitary adenoma
This is an open access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Pituitary adenomas arise from hormone-secreting cells of the anterior pituitary gland. The presenting symptoms are due to either excess or deficiency of pituitary hormones or local space-occupying effects. Loss-of-function mutations in the aryl hydrocarbon receptor-interacting protein gene (AIP) predispose to an autosomal dominant disorder with incomplete penetrance (20%–23%) associated usually with growth hormone-secreting pituitary adenomas leading to acromegaly or gigantism.1–3 To date, more than 100 different AIP variants have been identified, with the majority (75%) resulting in a missing or truncated AIP protein.2 3 A change in amino acid sequence due to missense variants could affect protein folding and stability4 and may alter the availability of protein–protein interaction sites. The C-terminus of AIP includes conserved tetratricopeptide repeat (TPR) domains, and alterations in key amino acids are known to disrupt secondary structure, leading to unstable proteins.5–7 While pathogenicity is beyond doubt for the truncating mutations, establishment of pathogenicity for missense variants can be challenging, posing therefore a key question for clinical genetic counselling and decision making.8
The strategies employed to establish pathogenicity for heterozygous tumour suppressor genes, such as AIP, include: allele frequency in the general population, loss of the wild-type (wt) allele in the tumour (loss of heterozygosity (LOH)), in silico prediction pipelines,9 in vitro functional studies and evaluation of variant segregation with the phenotype in large pedigrees.10 LOH analysis of tumourous tissue has also been exploited to determine the pathogenic role of AIP variants,11 12 and AIP immunostaining is significantly reduced in the majority but not in all patients carrying AIP mutations.13–15 In vitro functional studies have also been employed to evaluate the protein stability of AIP variants,4 their effect on cell proliferation13 and their interaction with PDE4A513 16 and RET,17 but these assessments are necessarily indirect.
However, the in vivo consequences of AIP missense variants have never been investigated. We aimed to develop an in vivo strategy to help determine the pathogenicity of missense AIP variants.
Materials and methods
Fly stocks and genetics
The Drosophila melanogaster strains used in this study: wiso (gift from Nic Tapon, London, UK), yw;Bl/CyO (Lindsley and Zimm),18 w* P{EP}CG1847G1839 (Bloomington Drosophila Stock Center: Stock ID: 32600),19 yw;; Ki, pp, Δ2–3, P{CaryP}attP40 embryos (BestGene Inc, California, USA) and yw; Act-Gal4/CyO.
Drosophila husbandry
Fly crosses were maintained at 25°C. For counting, the rescued males crosses were flipped every 9–10 days to prevent the mix of individual flies from different generations.
Generation of mutant CG1847 flies: imprecise excision screen
The CG1847 gene was mutated by P-element transposase-mediated deletion of genomic DNA. For this, a fly line was obtained, in which a P-element is inserted within the 5′UTR of CG1847: w*P{EP}CG1847G1839 (Bloomington Drosophila Stock Center).20 Females homozygous for the CG1847 mutation are not viable, while heterozygous mutant females develop normally. The resulting stocks were screened by PCR, and the putative mutants were identified via Sanger sequencing. Sequence chromatograms were visualised and analysed using the BioEdit Sequence Alignment Editor software (http://www.mbio.ncsu.edu/bioedit/bioedit.html) (Ibis Biosciences, Carlsbad, California, USA).
Rescue of CG1847 function
A genomic rescue construct containing the regulatory and coding regions of CG1847 (2763 bp) was generated, cloned into the pW@RpA vector (kindly provided by Professor Nick Brown’s laboratory, Cambridge, UK, details available on request).
To obtain the genomic rescue construct for hAIPwt, the AIP cDNA insert (1001 bp) was amplified from a pcDNA3-Myc-AIPwt vector.13 To obtain the genomic rescue construct of truncated AIP, the last 86 bp encoding for the seventh alpha helix were deleted. Transgenic lines for 11 different hAIP mutations (p.I13N, p.R16H, p.W73R, p.Q164R, p.C238Y, p.G272D, p.E293V p.A299V, p.R304Q, p.R314W and p.R325Q) were also generated. Mutagenic primers were designed using the Stratagene’s QuickChange Primer Design program at www.stratagene.com/qcprimerdesign. The QuickChange XL Site-Directed Mutagenesis kit (Agilent Technologies) was used, and mutagenesis was done according to standard recommended procedure.
All transgenic lines were generated by injecting the rescue constructs into Drosophila y1w67c23; P{CaryP}attP40 embryos, which enabled the generation of transgenic stocks with constructs on chromosome 2. These transgenic fruit flies stocks were balanced over the balancer chromosome CyO. For males resulting from the rescue crosses, the hAIP transgene (online supplementary figure 4B,C: middle panels) was detected using primers against human AIP cDNA. In addition, the presence of Y chromosome (bottom panels) was detected using a set of primers for the Ppr-Y gene.
Supplementary file 1
Statistical analysis
Experimental data sets were analysed in JMP (SAS institute). Statistical comparisons were analysed with one-way analysis of variance followed by a Tukey-Kramer test. Data are presented as mean ±SEM. A value of P<0.05 was considered to be statistically significant.
Western blotting analyses
The different UAS (Upstream Activation Sequence) insertions for the human AIP were confirmed to drive protein expression in combination with the actin-GAL4 using specific commercially available antibody. The Western blots were incubated overnight, at 4°C, with primary antibody anti-AIP/ARA9 Mouse Monoclonal21 (Novus Biologicals) at a dilution of 1:1000. Anti-Beta Tubulin, Mouse monoclonal (E7 Developmental Studies Hybridoma Bank)22 was used as a loading control at a dilution of 1:15 000. Secondary antibody IRDye 680 LT Goat anti-Mouse IgM (LI-COR Biotechnology) was used at a concentration of 1:1000. Odyssey Infrared Imaging System (LI-COR) was used for image acquisition. Results are representative of four independent western blot analyses from two independent experimental replicates.
Results
Characterisation of the Drosophila orthologue of human AIP
The D. melanogaster gene CG1847 (NM_132530.4)23 is the fruit fly’s single orthologue of human AIP. This three-exon gene is located on chromosome X at position 10F2, base pair (bp) 11 869 170 to 11 871 168 (Drosophila genome release August 2014). As it is located on the X chromosome, male flies will have only one copy of this gene.
CG1847 is a previously uncharacterised Drosophila gene, and no phenotypic fruit fly data are available in public databases. Protein alignment of CG1847 (FBtr0073567) and human AIP (hAIP, ENST00000279146) was performed by Clustal Omega24 and revealed a shared overall identity of 38% (Figure 1A). The three-dimensional theoretical model of CG1847 (Figure 1B) revealed a protein structure that closely resembles the published AIP protein structure , with a typical N-terminal peptidyl-prolyl cis-trans isomerases (PPIase)-like domain25 and the C-terminal TPR domains.26

Comparison of Drosophila CG1847 and human AIP proteins. (A) The human and Drosophila proteins are similar; they share 120 identical amino acids, 80 strongly conserved and 34 weakly conserved amino acids. Stars indicate identity, and colons and dots indicate high and low similarity amino-acids, respectively. (B) A three-dimensional predicted model of CG1847 indicates that the Drosophila protein has a similar structure to its human orthologue, with an N-terminal PPIase domain, three pairs of conserved antiparallel alpha-helices forming the tetratricopeptide repeat domains (TPRs) and the final extended α-helix, α−7. AIP, aryl hydrocarbon receptor-interacting protein gene.
CG1847 is an essential gene and its loss results in early lethality
We used imprecise P-element excision27 to generate a loss-of-function mutations in CG1847 (Figure 2A). One of the resulting lines harboured an excision of 2511 bp covering all three exons (CG1847exon1_3 , Figure 2A-ii). The deletion did not extend into the neighbouring genes CG2025 and CG11802, positioned 279 bp and 129 bp downstream of the CG1847 5′ and 3′ UTR regions, respectively. CG1847 is located on the X chromosome, and no viable hemizygous males were observed carrying this mutation. Because heterozygous females were normal, viable and fertile, this suggests that complete loss of CG1847 leads to lethality, similarly to mouse knockout models.28 29
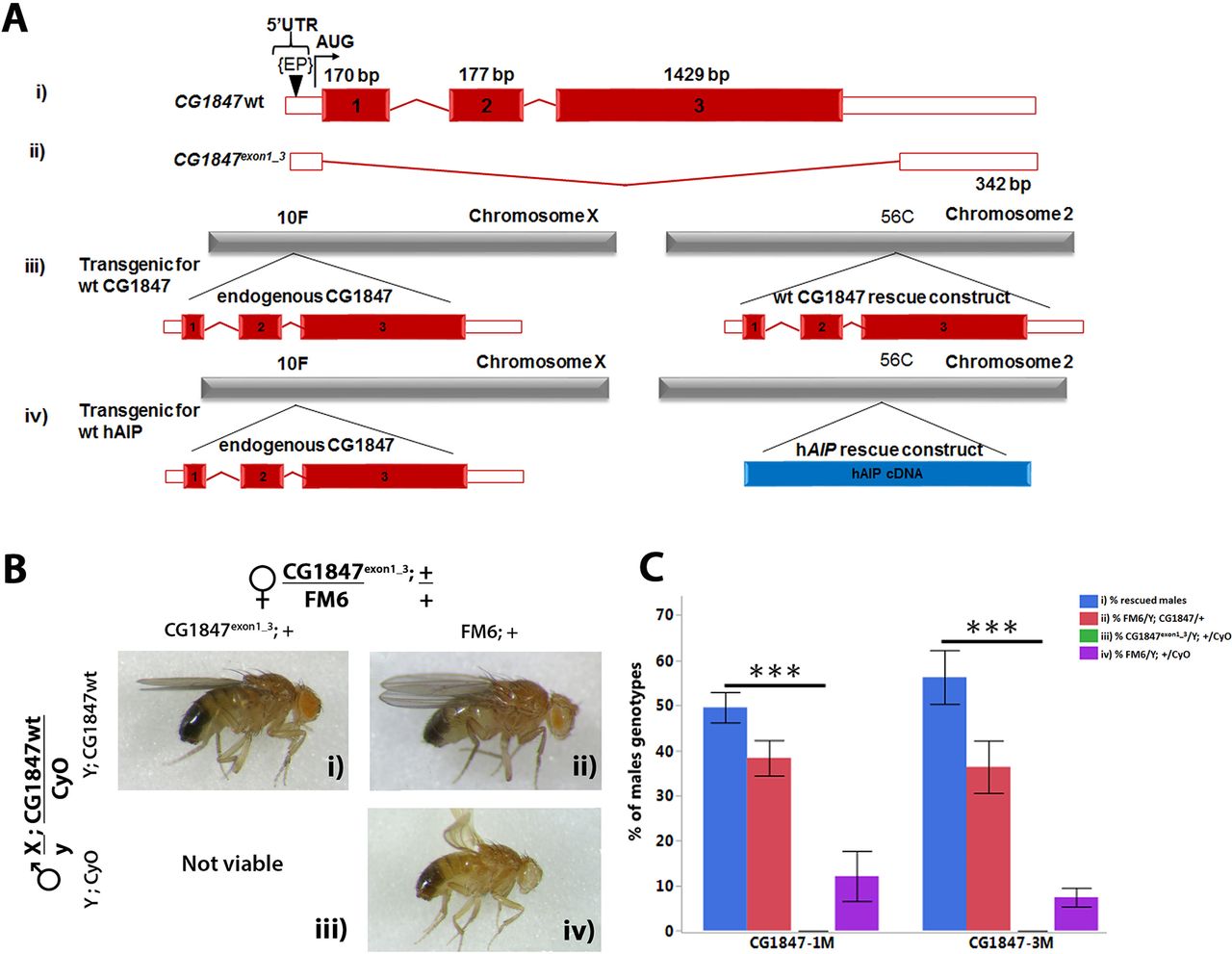
The lethality of CG1847 mutants can be rescued by expression of wild-type (wt) CG1847 under the control of its own promoter. (A) Schematic diagram of the wt CG1847 (i), CG1847exon1 _3 (ii), CG1847 rescue construct and (iii) transgenic generated human AIP rescue construct (iv). The diagram represent the mRNA, with red boxes representing coding region and white boxes representing 5′ and 3′ untranslated regions. (ii) The CG1847 mutant was generated by imprecise P-element excision of P-CG1847G1839 (EP in the figure). (iii) Schematic representation of transgenic animals with genomic rescue construct containing the regulatory and coding regions of CG1847 inserted on the second chromosome. (iv) Schematic representation of transgenic animals with the AIP cDNA rescue construct inserted on the second chromosome. (B) Transgenic males carrying the CG1847 genomic rescue construct and balanced over CyO were crossed to the CG1847exon1_3 heterozygous females and examined for their ability to rescue male lethality. Segregation of alleles and the possible combinations are shown in the lateral panels. Panel (i): male carrying the mutant CG1847 allele inherited from their mothers rescued by the wt CG1847 allele on the second chromosome. Panel (iii): males are not viable as they carry the CG1847 mutant allele and lack the genomic rescue construct from the paternal chromosome 2. Panels (ii) and (iv) depict male progeny lacking the CG1847 mutation. (C) Statistical analysis of rescue experiments with the CG1847 genomic rescue construct (n=4). The associated letters (panels i–iv) correspond to the phenotypes depicted in Figure 2B. X-axis labels CG1847-1M and CG1847-3M represent two different transgenic stocks carrying the rescue construct. Error bars represent SE of the mean. Asterisks indicate statistical significance as determined by Student’s t-test (**P<0.01). AIP, aryl hydrocarbon receptor-interacting protein gene; bp, base pairs.
To demonstrate that lethality of CG1847exon1_3 is solely due to the deletion of CG1847 coding sequence and not caused by additional mutations generated by the imprecise excision, transgenic flies carrying a genomic rescue construct on chromosome 2, containing the wt regulatory and coding regions of CG1847 (Figure 2A-iii), were generated and injected into Drosophila embryos (BestGene). Transgenic male flies were crossed with heterozygous CG1847exon1_3 mutant females (Figure 2B) to study the ability of the wt CG1847 rescue construct to reverse the lethality of CG1847exon1_3 males. Hemizygous mutant CG1847exon1_3 males expressing the wt CG1847 construct on chromosome 2 were viable (Figure 2B, panel i), suggesting that lethality of CG1847exon1_3 flies is indeed due to loss of CG1847. The degree of rescue was determined by analysing the proportion of each viable male genotype in the second generation (Figure 2C). All offspring without endogenous CG1847 or without the rescue construct (Figure 2B, panel iii) died at the larval stage. However, a small number of males without the rescue construct, but phenotypically similar to males in Figure 2B, panel iii, were viable due to a meiotic non-disjunction event in the previous generation, a low-frequency phenomenon common in Drosophila genetics (online supplementary material 1, figures 1–3). 30–33
Human AIP is able to functionally compensate for CG1847 loss
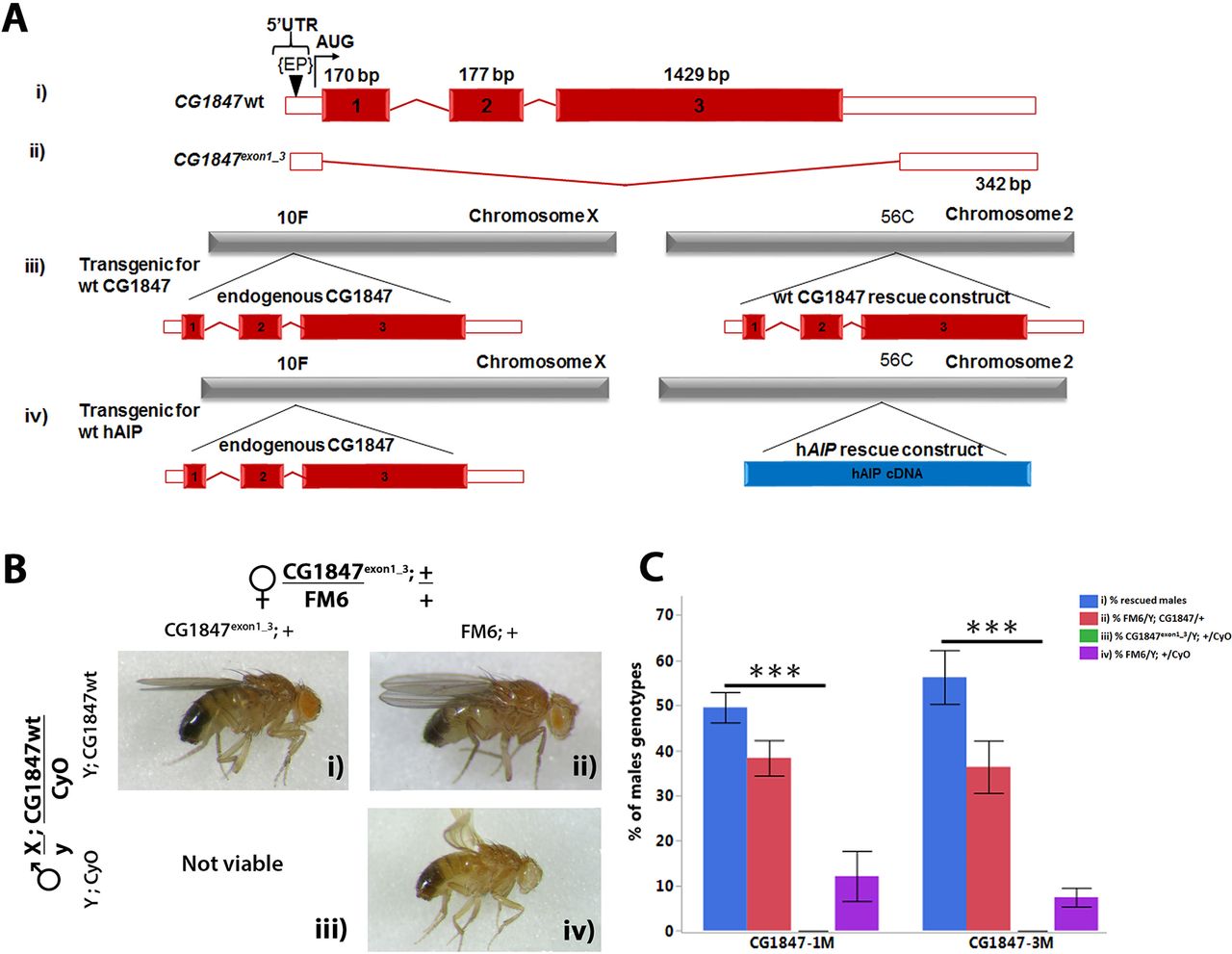
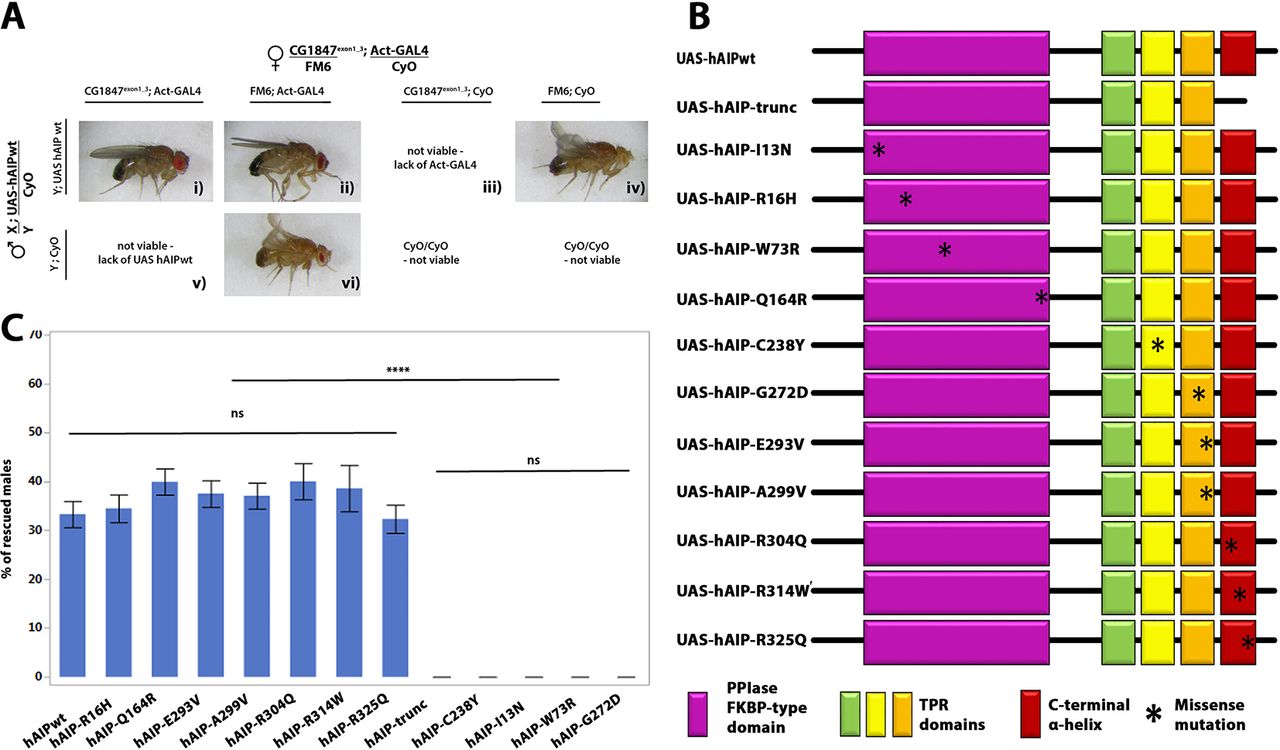
Since CG1847-deficient males die at an early larval stage, they could serve as a useful model to test the functional conservation between human and Drosophila AIP. We used the UAS-GAL4 system to express a human AIP (UAS-hAIP) transgene under the control of a ubiquitously active promoter (actin-Gal4) and assessed its ability to rescue the lethality of CG1847 mutant males. Ubiquitous expression of UAS-hAIP-wt was able to functionally replace CG1847 and rescue the lethality of Drosophila CG1847exon1_3 mutants (Figure 3A), confirming that AIP is functionally conserved between flies and humans. Moreover, the proportion of viable F1 males carrying the rescue hAIP construct was close to the expected proportion (33% observed vs 25% expected in case of full rescue, corresponding to 1 of 4 viable genotypes). Flies expressing the UAS-hAIP-wt construct could therefore be used as a positive control for testing hAIP variants with unknown significance identified in patients with pituitary adenomas. We also generated transgenic flies with a hAIP construct containing a truncation mutation of the last α-helix of the AIP protein—known to be crucial for AIP function26 34—and used this as a positive (pathogenic) control representing a non-functional AIP protein (Figure 3B). This truncated hAIP was unable to rescue the CG1847 exon1_3 mutant as no viable males were found expressing the truncated hAIP variant (Figure 3C).

Human AIP functionally complements the Drosophila orthologue. (A) Transgenic males with hAIP rescue construct were crossed to heterozygous CG1847 deficient females. The ubiquitous actin-Gal4 driver was used to drive the expression of the UAS-hAIP constructs during fly development. Panel (i) Images of F1 viable rescued males. Males expressing wt hAIP in the CG1847 mutant background (mutant CG1847 allele inherited from their maternal X chromosome and expression of the hAIP transgene on chromosome 2); panels (iii) and (v): males inheriting the mutant CG1847 allele and lacking hAIP expression are not viable. These two genotypes also serve as internal negative controls. Panels (ii) and (iv): males lacking the CG1847 mutation are viable. (B) Schematic diagram of UAS-hAIP constructs. Top, wt UAS-hAIP. Second line: artificially generated truncated hAIP lacking the seventh alpha helix. Transgenic lines for 11 different hAIP missense mutations (p.R16H, p.C238Y, p.A299V, p.R304Q, p.I13N, p.W73R, p.Q164R, p.G272D, p.E293V, p.R314W and p.R325Q) were also generated. The approximate position of amino acid changes introduced are indicated with an asterisk. Protein domains are indicated by the colour code shown below the deletion construct assembly (the colours of the domains match what is shown in the 3D model, Figure 1). (C) Quantitative analysis of in vivo rescue experiments using hAIP missense variants (n=6). Successful rescue of lethality was scored as the presence of males with the genotype CG1847exon1_3/Y; actin-Gal4/UAS-hAIP, which lacks endogenous CG1847. Error bars represent SE of the mean. Significant differences are indicated by asterisks (****P<0.0001). AIP, aryl hydrocarbon receptor-interacting protein gene; TPR, tetratricopeptide repeat; wt, wild type.
Human AIP missense variants differ in their ability to rescue CG1847 insufficiency
Having demonstrated that wt hAIP expression in CG1847 knockout flies is able to rescue male lethality, while a truncated version is not, we next tested the rescue ability of 11 hAIP missense variants (p.I13N, p.R16H, p.W73R, p.Q164R, p.C238Y, p.G272D, p.E293V, p.A299V, p.R304Q, p.R314W and p.R325Q), found as germline variants in patients with pituitary adenomas (Figure 3B).
Out of all these tested variants, we selected one known to be a relatively common polymorphism (p.R16H) as a negative control, and two variants, a truncation mutation and the p.C238Y missense pathogenic mutation, as positive controls. There are considerable amount of data showing that p.R16H is a benign variant; it does not segregate with the disease35 and various in silico predictions and functional studies (online supplementary material 2) suggest that it is a benign polymorphism.36 Truncating variants are known to be pathogenic, while the p.C238Y missense variant is also known to be pathogenic, based on segregation, in silico testing, in vitro functional studies (cell proliferation13 and the PDE4A5 binding assays16) and half-life data4 (online supplementary material 2).
Given that all transgenes were site-specifically integrated into the genome, they are predicted to be expressed at similar levels. Clinical and genetic data available for these missense variants are presented in online supplementary material 2.
For each missense variant, we compared the proportion of rescued males with the corresponding proportion obtained with the wt and truncated hAIP controls (Figure 3C). hAIP variants separated into two groups: the p.R16H, p.Q164R, p.E293V, p.A299V, p.R304Q, p.R314W and p.R325Q variants rescued the lethal CG1847exon1_3 phenotype at a similar rate as wt hAIP (Figure 3C). In contrast, four missense variants (p.C238Y, p.I13N, p.W73R and p.G272D) were unable to rescue the male lethality of CG1847 mutants (P=0.0001) similar to the truncated AIP variant. The data obtained using our in vivo model supports and strengthens the clinical and bioinformatics data indicating that p.C238Y, p.I13N, p.W73R and p.G272D AIP variants are pathogenic. However, our findings suggest that the p.R16H, p.Q164R, p.E293V, p.A299V, p.R304Q, p.R314W and p.R325Q variants are functionally normal sequence alterations in our experimental setting.
hAIP rescue constructs have equivalent expression levels
As ubiquitous expression of wt, p.R16H, p.Q164R, p.E293V, p.A299V, p.R304Q, p.R314W and p.R325Q hAIP resulted in rescue of CG1847exon1_3 mutant males, total protein was extracted from fly heads to perform western blot analysis. Analysis of hAIP protein in fly lysates revealed a 37 kDa band, equivalent to the band detected in human HEK293T cells used as positive control (figure 4). The various hAIP constructs show similar protein expression levels (figure 4). Normal flies (wiso) were used as a negative control to confirm that the AIP antibody we used does not detect the endogenous CG1847 protein. In addition, transgenic males carrying the UAS-hAIP constructs only, without a Gal4 driver, did not display a hAIP band, excluding any detectable ‘leakiness’ of the transgenic UAS constructs (online supplementary figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The different AIP constructs, wild-type human AIP (hAIP-wt) and various missense variants, show equivalent expression levels when ubiquitously expressed using the actin-Gal4 driver. CG1487 protein expression was not detected in wild-type flies (wiso), suggesting the anti-AIP antibody is specific for the human protein and does not recognise endogenous CG1487. β-tubulin was used as loading. Surviving males I13N*, W73R* and G272D* are viable as they have endogenous CG1487 but no hAIP due to non-disjunction (further details of the non-disjunction phenomenon is presented in online supplementary material 1). AIP, aryl hydrocarbon receptor-interacting protein gene.
While the p.R16H, p.Q164R, p.E293V, p.A299V, p.R304Q, p.R314W and p.R325Q hAIP missense constructs were able to rescue male lethality and displayed robust AIP expression (figure 4) similar to the level of wt hAIP, lysates from non-disjunction males (resulting from crosses with females carrying the p.I13N, p.W73R or p.G272D transgenes, figure 4, marked as *) did not contain human AIP protein. This confirmed that these males are indeed resulted from a non-disjunction event and are viable due to a normal copy of CG1847 (online supplementary figure 4).
Discussion
Patients with loss-of-function AIP mutations suffer from pituitary adenomas. For missense variants, the assessment of the functional and physiological consequences is hampered by the lack of robust in vitro and in vivo assays. In the era of increasing sequence data on human subjects, one of the most challenging issues is distinguishing pathogenic from non-pathogenic variants. In this study, combining a knockout for CG1847 with the Gal4/UAS binary expression system, we developed and optimised an in vivo system to ‘bioassay’ the pathogenicity of AIP variants found in patients with pituitary tumours.37
We have found that deletion of the CG1847 results in lethality, similarly to AIP knockout mouse models,28 29 and re-introduction of CG1847 rescued this phenotype, demonstrating the specificity of our KO model. We then exploited the experimental power of Drosophila genetics to evaluate the degree of functional conservation between human AIP and its fly orthologue by testing whether CG1847exon1_3 mutant flies could be rescued by human AIP.
A study of 287 human disease genes found a total of 178 (62%) genes having likely homologues in Drosophila. 38 Alignment of the human and fruit fly AIP proteins shows almost 40% identity, similar to the average level of protein identity between Drosophila and mammals.39
As the human gene was able to functionally compensate for the deletion of the Drosophila orthologue, our data support the evolutionary conservation of AIP gene function.
Furthermore, we have examined the effects of AIP variants in our Drosophila model in order to determine their pathogenicity. If a specific AIP variant rescues the lethality phenotype of CG1847exon1_3 mutant flies, this strongly suggests that the variant does not cause a major disruption of AIP function, at least with regard to Drosophila development. Conversely, failure to rescue the lethality phenotype indicates the variant is likely to be non-functional and could account for the human disease.
We investigated 11 AIP missense mutations identified in patients with pituitary adenoma. Summary of these mutations are listed in table 1 and further detailed in online supplementary material, part 1. Our negative and positive controls confirmed the validity of the assay; p.R16H rescued the lethality to a level similar to wt AIP, while the truncated and p.C238Y variants failed to do so.
Characterisation of AIP missense mutations identified in the various patients and investigated in this study
The lethality of CG1847exon1_3 mutants was rescued by 7 of the 11 tested hAIP missense variants (p.R16H, p.Q164R, p.E293V, p.A299V, p.304Q, p.R314W and p.R325Q), while four variants (p.C238Y, p.I13N, p.W73R and p.G272D) failed to do so. These data suggest that the latter four variants have a significant functional impairment or are unstable4 and therefore could represent pathogenic variants (see further discussion on these variants in online supplementary material 2). A similar strategy has previously been used to understand the conservation, functional role or importance of specific protein domains in human and Drosophila orthologues40–43 and could be employed to support clinical decision making.
Determining whether a variant is a disease-causing one is a significant challenge in the management of patients carrying a missense AIP variant,8 or indeed in any other partially penetrant disease.36 Evaluation of variant segregation with the phenotype in large pedigrees is the initial approach for investigating rare mutations.10 However, in case of AIP variants, this method is less practical due to the incomplete penetrance of the disease and the rarity of large families. Currently, there is no single method that can invariably predict the correct American College of Medical Genetics and Genomics (ACMG) category (‘pathogenic’, ‘likely pathogenic’, ‘uncertain significance’, ‘likely benign’ and ‘benign’) for missense variants. Although in silico prediction pipelines are often used, their results are not reliable, as shown recently, for endocrine genes including AIP.36 In addition to clinical data, such as segregation and variant frequency in the general population, various in vitro and in vivo methods could be used to help in the decision making. Inevitably, all methods have pitfalls. In vitro studies may not accurately recreate the environment present in a living organism. In the case of missense mutations, the change in amino acid sequence could disrupt their tertiary structure, with consequences for folding, stability and availability of protein–protein interaction sites. Robust and repeatable functional studies performed in clinical laboratories, however, have a significant role according to the ACMG guidelines.44
It was previously shown that the intact amino acid sequence of the TPR domains of AIP is essential for a proper interaction among amino acid residues in neighbouring alpha helices5; if these amino acids are changed by missense mutations, the resulted misfolded proteins are usually unstable.6 Various in vitro studies were employed to evaluate AIP variants, such as LOH analysis, splicing assays, cell proliferation, PDE4A5 binding and protein turnover.4 13 16 45 As we do not fully understand the mechanism of tumourigenesis induced by lack of AIP, the assay we might use in in vitro studies may not represent the true function of AIP. We demonstrated that AIP is an essential gene and several functional and comparative genomic analyses confirmed that essential genes are conserved during evolution.46–49 The fact that AIP is highly conserved among the species supports the fact that it is a disease-associated protein.50 As signalling pathways involved in organ development, cell proliferation, cell survival and cell migration are highly conserved in D. melanogaster,51 52 the results of fruit fly studies were shown to be transferable to humans; more than half of the known human disease genes, have homologues in fruit fly.53 We employed a Drosophila model organism to discover the conserved role of AIP and to avoid potential confounding factors arising from the redundancy and variability that can be generated by the analysis of more complex organisms. However, in vivo experiments, such as our Drosophila bioassay, may not correctly predict the functionality of a variant. Mouse studies, although closer to humans than Drosophila, can also provide misleading conclusions in some diseases.54 However, the fact that we used the human AIP protein in our studies and that this was sufficient to rescue the developmental function of the Drosophila orthologue is potentially a major advantage of our in vivo system, and this could be an additional approach to help clinicians reach the right conclusion.
In summary, we have engineered an in vivo bioassay for characterising patient-based AIP variants. The data presented support the evolutionary conservation of the AIP gene. Deletions of the endogenous Drosophila orthologue resulted in lethality of the flies, while the human gene can compensate for this loss. Rescue patterns of missense AIP variants can complement clinical and bioinformatics data and inform clinical decision making regarding AIP variants.8 16 The benefit of cascade genetic screening and clinical follow-up has already been established in AIP mutation-positive families,3 55 56 while family members in kindreds with non-pathogenic variants could be spared the psychological and financial burden of genetic testing and clinical follow-up.
Acknowledgments
We would like to thank to Professor Nick Brown (Department of Physiology, Development and Neuroscience, University of Cambridge) and Nic Tapon, Cancer Research UK for support regarding fly stocks and useful comments on the project. We are grateful for Professor Ashley Grossman for the careful reading of the manuscript. Fruit fly stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537). The authors would like to thank the ExomeAggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org about. The SNP annotations and genome location are accordingly Human Feb. 2009 (GRCh37/hg19) Assembly, Genome Browser website (http://genome.ucsc.edu).57
References
Footnotes
Contributors EDA and MK prepared the manuscript for publication, in consultation with PSR and RS. All other authors had a role in contributing intellectually to redrafting the manuscript. All experiments were performed by EDA with support from BK, CC, NM and AND. SR supported the statistical analysis and assisted with drafting the manuscript. CP created the CG1847 protein theoretical model. All authors have seen and approved the final manuscript.
Funding Grant number MR/M018539/1 granted by Medical Research Council; WS733753 granted by Pfizer UK, November 2014 Early Career Grant awarded by Society for Endocrinology; #2011-5 awarded by William Harvey Research Foundation.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Not required as no additional unpublished data from the study.