Article Text

Abstract
Parkinson’s disease (PD) is a complex and heterogeneous neurological condition characterised mainly by bradykinesia, resting tremor, rigidity and postural instability, symptoms that together comprise the parkinsonian syndrome. Non-motor symptoms preceding and following clinical onset are also helpful diagnostic markers revealing a widespread and progressive pathology. Many other neurological conditions also include parkinsonism as primary or secondary symptom, confounding their diagnosis and treatment. Although overall disease course and end-stage pathological examination single out these conditions, the significant overlaps suggest that they are part of a continuous disease spectrum. Recent genetic discoveries support this idea because mutations in a few genes (α-synuclein, LRRK2, tau) can cause partially overlapping pathologies. Additionally, mutations in causative genes and environmental toxins identify protein homeostasis and the mitochondria as key mediators of degeneration of dopaminergic circuits in the basal ganglia. The evolving mechanistic insight into the pathophysiology of PD and related conditions will contribute to the development of targeted and effective symptomatic treatments into disease-modifying therapies that will reduce the burden of these dreadful conditions.
- parkinson’s disease
- parkinsonism
- basal ganglia
- disease mechanisms
- genetics
Statistics from Altmetric.com
Introduction
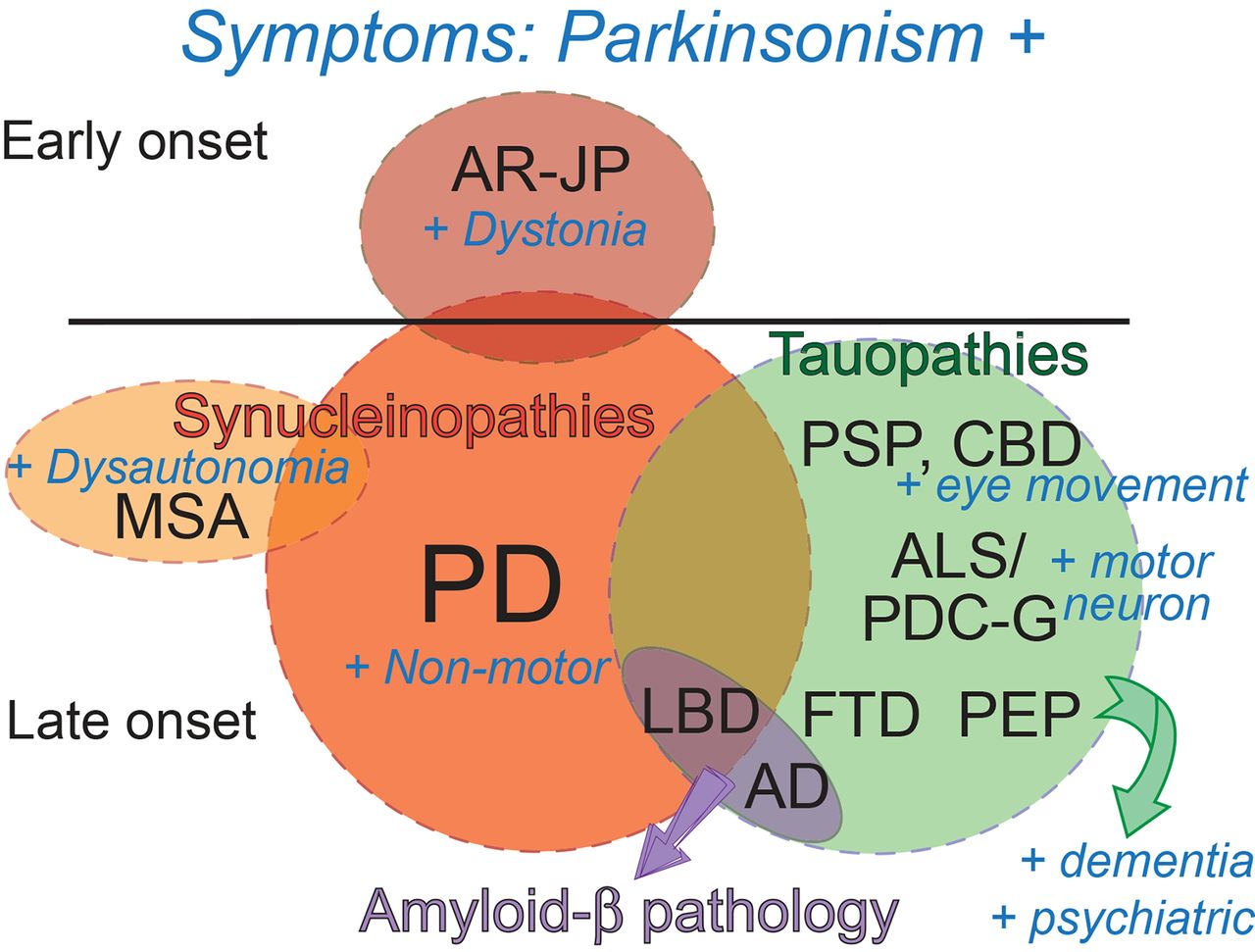
Parkinson’s disease (PD) is the most prevalent movement disorder and the second most common neurodegenerative condition after Alzheimer’s disease (AD).1 PD is a complex disorder with early non-motor symptoms, core motor symptoms, and late cognitive and psychiatric complications.2 The core motor symptoms of PD—bradykinesia (slow movements), muscular rigidity, resting tremor, and abnormal posture and gait—were recognised in the original description of the disease 200 years ago by J Parkinson,3 which JM Charcot later used to define the disease.4 The combined classical motor disturbances of PD are referred to as parkinsonism or parkinsonian syndrome. Although PD is the major cause of parkinsonism, these features are shared by many conditions, including AD and others with strong overlap with PD at the time of diagnosis. In addition, PD is heterogeneous and can progress mainly as a movement disorder or develop cognitive and psychiatric symptoms that overlap with several dementias.2 The main pathology of PD are protein aggregates enriched in α-synuclein (α-syn) called Lewy bodies (LB) that accumulate prominently in dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) and in other brain regions that correlate with late symptoms.5 6 This α-syn pathology is also shared by other PD-related conditions (synucleinopathies) with overlapping but distinct features (figure 1). Understanding the pathophysiology and genetics of PD and related disorders is a critical step to gain insight into the mechanism of DA neuron degeneration and design definitive disease-modifying therapies that will eliminate the burden of these common and dreadful conditions.

Symptom overlap among PD and related parkinsonian disorders. Parkinsonian disorders are represented as circles (not at scale) and the symptomatic overlap is shown. Synucleinopathies are shown in red tones, tauopathies are shown in green and Aβ pathology is shown in purple. The symptoms are shown in blue. All these conditions share the parkinsonism, and under each disease the additional symptoms (+) are shown. LBD is a unique condition that can present with LB or Aβ pathology and has prominent PD-like symptoms+dementia and psychiatric disturbances; thus, LBD can be considered a mixed PD/AD pathology and is placed in the overlapping area. Aβ, amyloid-β; AD, Alzheimer’s disease; ALS/PDC-G, amyotrophic lateral sclerosis/parkinsonism with dementia complex of Guam; AR-JP, autosomal recessive juvenile PD; CBD, corticobasal degeneration; FTD, frontotemporal dementia; LB, Lewy body; LBD, Lewy body dementia; MSA, multiple systems atrophy; PD, Parkinson’s disease; PEP, postencephalitic parkinsonism; PSP, progressive supranuclear palsy.
Clinical features
Although the overall incidence of PD is around 15 per 100 000 person-years, age is the most important risk factor, with peak incidence around 80 years old.1 PD grows exponentially with age, reaching an incidence of 1% at age 65 and 5% by age 85.1 Continuous medical advances and the increase in life expectancy worldwide project the doubling of cases of PD by 2030, which will cause a serious public health burden. In addition to age, gender and race also contribute to the risk of PD, with men, Hispanics and non-Hispanic whites showing the highest incidence.7
PD is the most common condition presenting with the core motor symptoms of parkinsonism. These symptoms are caused by the loss of dopamine-producing neurons in the SNc that project to the putamen.5 6 The basal ganglia contains several nuclei with a critical role in the control of voluntary movements. Dopamine-dependent disinhibition through the direct and indirect pathways activates thalamic nuclei that stimulate the motor cortex to initiate voluntary movements. The loss of dopamine in the SNc impairs disinhibition, preventing the stimulation of the motor cortex. The early prominent degeneration of SNc and functioning basal ganglia explains the response of patients with PD to dopamine replacement therapy in the form of L-dopa.2 8 Patients with PD also respond to direct stimulation of different basal ganglia nuclei via deep brain stimulation (DBS).9 The robust symptomatic improvement of parkinsonism to these two treatments corroborates current anatomical and physiological understanding of PD pathology. However, these therapies do not halt or slow down the underlying pathology, which progressively destroys the SNc, the basal ganglia and later spreads to other brain regions.5 6 10
In addition to the well-known motor dysfunction, PD is characterised by non-motor symptoms affecting the brain and the peripheral nervous system.11 Constipation, sleep perturbations and hyposmia (loss of smell sense) typically precede the onset of the main motor symptoms by almost two decades.11 These symptoms do not respond to L-dopa or DBS, uncovering the pathology of multiple independent foci. Progression of the non-motor symptoms can lead to incontinence, psychosis and dementia,12 13 painting a complex picture of the late-stage pathophysiology of PD with significant overlap with other conditions.
Pathology
The loss of DA neurons in the SNc is accompanied by LB pathology,14 which consists of protein aggregates enriched in α-syn. In terminal stages, this LB pathology is found throughout the brain, nerve cord and peripheral neurons.5 Interestingly, LB pathology has been proposed to propagate through the brain in a stereotypic pattern that correlates with the progression of symptoms.15 This propagation is likely mediated by cell-to-cell transmission of aberrant protein conformations similar to the templated conversion of the prion protein. The mechanism of this prion-like transmission16–19 is proposed to be conserved in other amyloid proteins responsible for many neurological conditions, including the amyloid-β (Aβ) peptide and tau linked to AD.
α-Syn is a 140-amino acid-long intrinsically disordered protein highly expressed in brain neurons, mainly in the nucleus and in presynaptic terminals.20 21 α-Syn can misfold and aggregate into soluble oligomers and protofibers that can progress to insoluble fibres visualised as LB.22 23 The soluble α-syn assemblies have been shown to be more toxic than the fibres in experimental settings, suggesting that the LB acts as sinks for aberrant proteins and thus have a protective role.24–26 Although α-syn seems to play a central role in the pathogenesis of PD and other related synucleinopathies, it is not the only agent causing parkinsonism.
Environment versus genetics
It is clear that PD has a strong environmental component. Multiple studies have confirmed the higher risk of developing PD in populations exposed to heavy metals (well-water drinking) and pesticides.27 Rotenone and paraquat are two pesticides that disrupt the respiratory chain in the mitochondria and cause oxidative stress.28 29 This connection was strengthened when in 1983 the use of heroin contaminated with methyl-phenyl-tetrahydropyridine (MPTP) led to parkinsonism in young adults.30 MPTP is metabolised into 1-methyl-4-phenylpyridinium (MPP+), which induces similar mitochondria toxicity as rotenone and paraquat, supporting the contribution of environmental toxins to PD. An interesting report linked parkinsonism in the island of Guam to the consumption of seeds from the native cycad fruit,31 although this association seems to be weaker now, perhaps due to increased awareness. Additionally, head injury and some antipsychotics enhance the risk for PD,2 exposing the delicate balance that neuroinflammation and neurotransmission play in brain disease.
The strong influence of environmental factors in PD, the age dependence and the variable disease presentation, along with the incomplete penetrance and variable expressivity of several PD genes, has traditionally lowered the estimation of its heritability (1%–5%). Recent epidemiological studies have reported up to 10%–30% heritability and new PD genes have been identified, supporting the strong influence of genetics in PD.32 33 In fact, the formal distinction between familial and sporadic PD needs to be revisited since several PD-causing mutations are present in cases lacking family history. This can be explained by incomplete penetrance of some mutations, early death of relatives before exhibiting symptoms or inaccurate medical records. Here, we describe the monogenic forms of PD and related parkinsonian disorders. Understanding the mechanisms linking these causative and susceptibility genes can improve diagnosis and better predict the prognosis of these disorders. We further discuss a model of a continuous disease spectrum that explains the overlap of symptoms, genetics and/or pathology.
Autosomal dominant PD
Following the discovery of α-syn presence in LB,23 the A53T missense mutation in α-syn (encoded by SNCA) was identified in 1997 in a family showing autosomal dominant inheritance of PD.34 Later studies identified other missense mutations in α-syn (A30P, E46K, H50Q, A53E) that confirm the causative role of α-syn in PD.35 These mutations have high penetrance and are associated with early-onset PD in the 40s or 50s with typical motor symptoms and good response to L-dopa. These patients show rapid disease progression, early dementia and widespread distribution of LB pathology. A recent report described a fourth missense mutation in α-syn—G51D—associated with parkinsonism-pyramidal syndrome in a French family.36 Furthermore, some families can also carry low penetrant duplications or triplication of wild-type SNCA with typical PD motor and non-motor symptoms. Recent unbiased genome-wide association studies (GWAS) in European and Japanese populations37 38 suggest the presence of non-coding variants in the SNCA locus (mainly in the 3’ end) proposed to regulate gene expression. So far, the evidence points to a gain-of-function (GOF) mechanism for α-syn triggering PD. This model is consistent with other brain proteinopathies that share pathogenic mechanisms linked to oligomer neurotoxicity and cell-to-cell transmission.
Despite the central role of α-syn in PD, mutations in leucine-rich repeat kinase 2 (LRRK2) are the major cause of familial PD.39 40 Subsequent work has identified around 50 mutations in LRRK2 associated with both familial and sporadic forms of PD,41 with frequencies that can reach up to 40% in some populations. These mutations confer increased risk of PD, with modest effects in some cases due to low penetrance. Highly penetrant mutations (eg, G2019S) are pathogenic in laboratory models, linking LRRK2 dysfunction to PD. Patients carrying LRRK2 mutations present with typical late-onset PD symptoms, including non-motor symptoms, LB pathology and degeneration of DA neurons in the SNc; however, asymptomatic 80-year-old carriers of the G2019S mutation have been described.41 LRRK2 is a complex gene comprised of 51 exons encoding a broadly expressed 2200-amino acid-long protein with kinase activity.39 40 42 The most common mutations in LRRK2 seem to affect the kinase activity and G2019S increases autophosphorylation, suggesting GOF pathogenic mechanisms.43
Glucocerebrosidase (GBA) encodes a lysosomal protein that degrades glucocerebroside. Mutations in GBA lead to the autosomal recessive Gaucher’s disease, a lysosomal storage disorder.44 45 Interestingly, some patients with Gaucher’s disease seem to develop parkinsonian symptoms, and heterozygous relatives have 6-fold to 10-fold increased risk of developing PD.46 Only about 25% of carriers of GBA mutations have a family history of PD, with the rest contributing to ‘sporadic’ PD. These patients show clinical symptoms and pathology typically found in idiopathic PD, except for a younger age of onset.
In addition, three more recent genes have been identified with a minor contribution to genetic PD. Mutations in vacuolar sorting protein 35 (VPS35) are associated with late-onset PD,47 48 and importantly VPS35 mutations are also found in ‘sporadic’ PD cases.33 Only two mutations have been characterised in patients, with D620N representing a hot spot. The VPS35 protein seems to shuttle cargo between mitochondria and lysosomes, and VPS35 mutants cause mitochondria fragmentation.49 Mutations in DNAJC13 have only been identified in a mixed Saskatchewan (Canada) population and are present in both familial and sporadic cases.47 50 DNAJC13 encodes a molecular chaperone of the Hsp40 family that participates in receptor-mediated endocytosis and localises to the lysosome. Mutations in CHCHD2 were identified in Japan.51 Three variants have been identified in patients, suggesting a causative link, although little is known about this gene. Together, the old and new PD genes provide a more complete understanding of the genetic landscape of PD and confirm the contribution of low penetrant mutations to the underestimation of the genetic burden in PD.
Autosomal recessive juvenile PD
Some familial forms of PD with autosomal recessive inheritance present very early onset (‘juvenile’) and show both typical and atypical PD symptoms. In 1998, mutations in parkin were identified in Japanese families,52 and now represent the major cause of young-onset parkinsonism.53 These patients present with typical motor symptoms and good response to L-dopa, but also slow-course, prominent dystonia, DA neuron loss in the SNc with sporadic LB and absence of non-motor symptoms (figure 1). These differences suggest a more restricted pathology to the SNc and basal ganglia with minor contribution of α-syn pathology.54 55 Parkin encodes a widely expressed E3 ubiquitin ligase, a key regulator of protein degradation by the proteasome, suggesting a mechanistic connection with GBA. The FBXO7 (PARK15) gene encodes another E3 ubiquitin ligase implicated in autosomal recessive juvenile PD. Mutations in FBXO7 are less common than those in parkin, disease progresses more rapidly, and symptoms include dementia, dystonia and motor neuron degeneration.56
Mutations in PTEN-induced kinase 1 (PINK1) and Daisuke-Junko-1 (DJ-1) cause diseases similar to that of parkin mutations, with typical motor symptoms that appear very early, slow progression and lack of non-motor symptoms.57 58 However, their pathology is unknown since no autopsies have been conducted, so far. PINK1 is a serine-threonine kinase that localises to the mitochondria. DJ-1 is a 183-amino acid protein of unknown function localised in the mitochondria following oxidative stress.59 Parkin, FBXO7, PINK1 and DJ-1 may exert neuroprotective functions under physiological conditions, and their loss seems to trigger specific degeneration of vulnerable DA neurons. These genetic discoveries support key roles of disrupted protein and mitochondria homeostasis in triggering the degeneration of DA neurons (see below).
Other synucleinopathies
LBs are also prominent in at least two other parkinsonian disorders. Lewy body dementia (LBD) or dementia with LBs presents with typical parkinsonism, non-motor symptoms, early dementia and psychiatric disturbances, including visual hallucinations, aggression and depression.60 In many regards, LBD appears as PD with early dementia. LBD can present with prominent LB or Aβ pathology, making this a PD/AD mixed pathology (figure 1). As expected, the LB pathology appears in brain areas associated with motor and cognitive symptoms. Most LBD cases are sporadic, but rare autosomal dominant forms of LBD are associated with mutations in SNCA and LRRK2,60 the most prominent PD-causative mutations. GBA and ApoE4, risk factors for PD and AD, respectively, also increase the risk of LBD.60 Overall, the pathological and genetic similarities between PD and LBD suggest that these clinical entities belong to a disease continuum that features several other conditions (figure 2).

{kind=link}
{kind=link}
Visualisation of PD-related spectrum in a continuous arch. PD-related pathologies are arranged in an arch (orange) that shows subtle transitions in clinical symptoms. MSA is placed at one end of the spectrum because it is a unique systemic synucleinopathy with glial pathology. AD occupies the other end of the spectrum next to other dementias because parkinsonism is less common. The dominant protein pathology (α-syn or tau) is shown in the middle arch. In the centre (grey), the impact of age is shown, which is a strong determinant in all diseases, except for AR-JP. See figure 1 for abbreviations. α-syn, α-synuclein; MPTP, methyl-phenyl-tetrahydropyridine; Prot., protein.
Multiple systems atrophy (MSA) is a synucleinopathy with parkinsonism and additional clinical features very distinct from PD (figure 1). MSA is a sporadic, multisystem, rapidly progressive condition characterised by prominent failure of the autonomic nervous system or dysautonomia (incontinence, orthostatic intolerance), and cerebellar dysfunction (ataxia).10 Patients with MSA show neuronal loss in the striatonigral and olivopontocerebellar structures that explain the parkinsonism and ataxia. But the main pathological hallmark of MSA is the presence of LB pathology in oligodendrocytes, leading to demyelination of many neuronal tracts. Patients with MSA do not respond to dopamine replacement therapy, indicating a distinct underlying pathology that spares the DA neurons in the SNc. MSA fits in one end of the PD-dementia spectrum due to several unique aspects (figure 2).
Parkinsonism with tau pathology
Parkinsonism can also be present in conditions characterised by prominent tau pathology (figure 1). Tau pathology appears in the form of the neurofibrillary tangles (NFT) that contain insoluble and hyperphosphorylated forms of the microtubule binding protein tau.61 The tauopathies with parkinsonism can be divided into three subclasses: conditions similar to PD at onset, dementias with secondary parkinsonism and environmental exposures. Corticobasal degeneration (CBD or corticobasal ganglionic degeneration) and progressive supranuclear palsy (PSP) are two tauopathies with partial symptomatic overlap with PD. But, as these diseases progress, they are more easily distinguished from PD. Patients with CBD suffer progressive degeneration of the cortex and the basal ganglia leading to cognitive and psychiatric symptoms combined with asymmetric parkinsonian features. In addition, these patients can present characteristic movement disturbances—dystonia, perturbations in eye movement, apraxia (loss of speech) and loss of inhibition and empathy. PSP can have an overlapping presentation with CBD plus prominent supranuclear gaze palsy and loss of motivation and interest. NFT accumulates in basal ganglia, medulla and other deep nuclei consistent with these symptoms. CBD and PSP display both neuronal and astrocytic NFT, a less common glial pathology in proteinopathies. Despite the symptomatic overlap with PD, neither patients with CBD nor patients with PSP respond well to dopamine replacement therapy, consistent with degeneration of the basal ganglia, which contains the target neurons for dopamine input. Both CBD and PSP are mainly sporadic and accumulate a particular tau isoform (4R). But PSP can be associated with mutations in tau and LRRK2.
The tauopathies with secondary parkinsonism include frontotemporal dementia (FTD) and AD,62 in which parkinsonism is a consequence of the late-stage spread of pathology (figure 1). Although tau pathology is critical in AD, AD seems to be triggered by Aβ oligomers and tau mutations never cause AD.63 However, tau mutations can cause some forms of the complex FTDs, namely frontotemporal dementia with parkinsonism in chromosome 17. The complex genetics and pathology of FTD will not be discussed in further detail here.64 65 These two conditions are placed at the other end of the continuum of the parkinsonian disorders due to their lower overlap with PD (figure 2).
Interestingly, two tauopathies with parkinsonism are caused by environmental factors (figure 1). Although no longer prevalent, influenza infections can cause postencephalitic parkinsonism with tau pathology in the SNc.66 Amyotrophic lateral sclerosis/parkinsonism with dementia complex of Guam is linked to the consumption of the cycad fruit in the Pacific island of Guam.31 However, the specific toxin responsible for this pathology has not been clearly identified and the disease has almost disappeared in recent years. These environmental links are important because they highlight the high vulnerability of these circuits and may contribute to the identification of the cellular mechanisms triggering these complex pathologies.
Other genes implicated in PD
In addition to the causative PD genes, other loci have small effect, strong statistical association and replication in multiple ethnicities. So far, no pathogenic mutations have been identified in these loci that can elucidate their causative roles. PARK16 and BST1 contain two variants each with strong association to PD in both Japanese and European populations.37 PARK16 is a complex locus containing five genes that have been confirmed in several studies. BST1 (bone marrow stromal cell antigen 1) was identified originally in Japanese populations, but recent studies have shown association also in European populations,67 and its role in PD may be mediated by dysregulating intracellular calcium.
Access to genomic technologies has led to hundreds of GWAS and the identification of thousands of PD susceptibility loci. However, the lack of replication for most candidates caused significant distraction from the key need of dissecting the central molecular mechanisms mediating PD.68 A recent meta-analysis of >800 GWAS validated the known PD loci described above and identified eight additional loci with modest effect but robust association in multiple ethnicities: ACMSD/THEMI63, CCDC62/HIP1R, DGKQ/GAK, HLA, ITGA8, MCCC1/LAMP3, STK39 and SYT1I/RAB25.67 The polymorphisms with the largest effects (measured by odds ratio (OD)) correspond to GBA in Caucasian populations (OD 3.5), LRRK2 in Asian populations (OD 2.33) and SYT11/RAB25 in Caucasians (OD 1.73). The rest of the loci showed ODs between 1.10 and 1.35, the expected low risk for multigenic diseases with environmental contribution.67 However, there is no direct evidence yet for the involvement of these susceptibility genes in the causation of PD. Having just a handful of candidates instead of thousands will facilitate the search for variants affecting their coding sequence, splicing or expression. Furthermore, model systems can be exploited to determine their physical or functional interaction with core PD genes. It is likely that genetic and environmental context may critically impact the expressivity of some loci, whereby only certain genetic combinations and environmental factors trigger disease. Demonstrating these interactions experimentally will require computational modelling approaches with the potential to (1) dissect disease mechanisms and (2) predict both disease risk and prognosis.
From genetics to molecular mechanisms of parkinsonism
The identification of genes and environmental toxins causative of the parkinsonian syndrome provides a starting point to dissect the molecular mechanisms responsible for pathogenesis. Understanding these mechanisms and the site of action of the pathogenic genes and toxins is critical for designing disease-modifying therapies. We highlight here three mechanisms with a prominent role in PD/parkinsonism: protein dyshomeostasis, mitochondria dysfunction and selective vulnerability.
Protein dyshomeostasis
PD and other forms of parkinsonism can present with LB, tau and other protein pathologies. Oligomeric assemblies of misfolded α-syn demonstrate toxicity in cell culture, but no specific species (multimer) has been identified as the most toxic.24 26 Overexpression of α-syn is toxic in yeast, Caenorhabditis elegans and Drosophila, supporting the disease-triggering role of α-syn.69–72 Moreover, viral expression of α-syn induces degeneration of DA neurons in rats and primates, directly linking α-syn to parkinsonism.73–75 Furthermore, the LB pathology present in asymptomatic elderly brains may represent a slow-progressing, presymptomatic disease state. Alternatively, LBs may represent a protective mechanism to reduce the concentration of highly toxic, soluble α-syn oligomers.24 26 Moreover, LRRK2 knockout suppresses LB pathology in mice and rats, suggesting that LRRK2 negatively regulates α-syn clearance in microglia.76–78 Other factors that may trigger pathology with or without α-syn pathology are alterations in cellular proteostasis, including GBA mutations that perturb the lysosome and mutations in the E3 ubiquitin ligases parkin and FBXO3. Interestingly, parkin overexpression rescues α-syn toxicity in a rat viral model, functionally linking these two PD genes.79 Finally, age is the most critical risk factor in PD, and ageing is known to weaken proteostasis through decreased expression of molecular chaperones, co-chaperones and other critical factors for protein degradation.80 Overall, altered proteostasis is a critical cellular mechanism mediating parkinsonism.
Mitochondria
Several environmental toxins have a direct link to the mitochondria, including MPTP and the pesticides rotenone and paraquat. These toxins interfere with the respiratory chain and cause oxidative stress that ultimately kills DA neurons with such specificity that MPTP toxicity became the best mouse model of PD for some time. In addition to toxins, parkin and PINK1 mutants exhibit robust mitochondrial defects in Drosophila and mammalian cells due to perturbations in mitochondria fusion and fission.81–84 Moreover, parkin overexpression rescues PINK1 phenotypes, suggesting related physiological functions. Additionally, FBXO7 and Parkin interact to mediate mitophagy,85 while FBXO7 overexpression rescues parkin loss-of-function in Drosophila, suggesting the functional overlap between these two E3 ubiquitin ligases.56 However, mutations in parkin and PINK1 show no phenotypes or functional interactions in mice, which could be due to genetic redundancy.86 DJ-1 has been shown to translocate to mitochondria to protect against oxidative stress, and seems to play a key regulatory role against oxidative stress by stabilising the antioxidant master regulator nuclear factor erythroid 2-related factor 2.87 VPS35 mutations alter mitochondria function and cause mitochondria fragmentation.88 Overall, both toxins and genetics disrupt the mitochondria and the DA system.
Selective vulnerability
Several protein pathologies and mitochondria dysfunction disrupt DA circuits, highlighting their heightened vulnerability. Understanding this selective vulnerability will provide critical cues to guide the development of therapies to support the survival of these neurons. Two important aspects of selective vulnerability in PD are the development of LB pathology and selective neuronal death. The LB pathology is proposed to spread synaptically through established networks by both anterograde and retrograde mechanisms. However, not all neurons in these networks accumulate LB, suggesting differences in the ability to either uptake α-syn seeds or degrade them before accumulating in fibrillar conformations. α-Syn is a highly expressed protein throughout the brain, and expression differences between neurons have not been suggested to play a role in selective vulnerability.89 The factors mediating these differences are currently unknown since the prion-like spread models are still under investigation.16–19
Regarding selective cell death, around disease onset, patients with PD already show loss of 10%–20% of DA neurons consistent with the distribution of LB pathology. As disease progresses, many areas accumulate LB pathology but show no significant cell death. Thus, cell autonomous factors appear to modulate the vulnerability of neurons within the PD network. Recent work proposes that DA neurons in the SNc have a distinctive physiology that arises from the production of slow rhythmic action potentials in the absence of excitatory input, which is critical for the broad neuromodulatory activity of the DA network.89 This autonomous rhythmic activity creates large oscillations in intracellular calcium concentrations that maintain the action potentials and stimulate the mitochondria to produce the needed energy. Since DA neurons are known to have low calcium-buffering activity (eg, low calbindin),90 high intracellular calcium ends up inducing oxidative stress, damaging mitochondria, promoting α-syn aggregation and inducing apoptosis.89 Thus, vulnerable neurons seem to be in a ‘bioenergetic and proteostatic tipping point’89 that can be easily disrupted by genetic variants, exposure to exogenous insults and ageing.
Concluding remarks and unanswered questions
The heterogeneity and overlapping clinical manifestations, pathology and genetics of PD-related pathologies (figure 1) lend support to the conclusion that they are part of a disease continuum: from classic PD (no dementia) to pure AD (no parkinsonism) with several mixed conditions in between (figure 2). This continuum of symptoms together with the lack of helpful biomarkers and diagnostic imaging explains the challenges of diagnosing at onset, which is critical for prescribing targeted symptomatic treatments (L-dopa?) and disease prognosis. On the other hand, the common features of these conditions underscore the opportunity of identifying shared pathogenic mechanism that could lead to the development of disease-modifying and symptomatic therapies with broad benefits. Recent advances have revealed a stronger genetic contribution in PD than previously thought, uncovering the role of several PD genes in sporadic disease associated with low penetrant mutations. The interplay between genetics, environmental toxins and selective neuronal vulnerability is proposed to disrupt cellular proteostasis and mitochondria dynamics. These perturbations then launch the pathogenic cascade in DA circuits that, over decades, spreads through a network of vulnerable neurons that result in the complex clinical picture of the parkinsonian syndrome.
Recent developments in AD immunotherapeutics support the potential of turning the immune system against brain proteinopathies.91–93 Active immunotherapy (vaccination against Aβ42) showed a promising reduction in brain Aβ42 and revealed the potential dangers of an over-reacting immune system.94 Passive AD immunotherapy (direct administration of an antibody) with lineal and conformational antibodies against Aβ42 has failed to show significant cognitive protection in symptomatic patients.93 Still, trials are ongoing with the hope that the most promising antibodies will work in presymptomatic and at-risk patients.95 The experience accumulated over the last 15 years in AD immunotherapies can be translated to other related proteinopathies, including α-syn and tau. Despite the dangers of vaccinating against an endogenous protein, active vaccination has the potential advantage of an easy administration with sustained, long-term benefits. The approach against α-syn consists of vaccination with short peptides that mimic α-syn domains but lack sequence homology to avoid tolerance to self-proteins.96 Following supportive preclinical data in several mouse models, two α-syn vaccines showed good tolerability and good immune response in patients with PD and MSA,96 97 although no supportive clinical data are available yet. On the other hand, passive immunotherapy offers greater control against adverse effects and enables antibody engineering for increased brain penetration, stability and lessened cellular response. Several N-terminal and C-terminal antibodies have produced exciting results by clearing α-syn, reducing its propagation and lessening motor dysfunction.91 Safety data are positive for one phase I trial with a C-terminal antibody (PRX002), which also shows significant reduction of serum-free α-syn. A larger study with this antibody and with a second antibody is ongoing, but no efficacy data are still available.97 Based on the lessons from AD immunotherapy, patients in the prodromal phase of PD, LDB and MSA, and carriers of familial mutations, should be the ideal targets in these trials because they can be treated while a significant number of cells can be protected from further degeneration.
Despite these remarkable advances, many critical questions remain to be elucidated. (1) The exact mechanisms for the initiation of the pathology are unknown, a problem exacerbated by the start of pathology 10–20 years before clinical diagnosis. A combination of external stressors (toxins) and genetic susceptibility may explain the heterogeneity of onset and progression in idiopathic PD, but the exact combination of the causative factors is unknown. (2) α-syn and tau have prominent roles in several diseases, but the factors triggering neuronal (eg, PD) versus glial (eg, MSA) pathologies are unknown. (3) The genotype/phenotype correlation is poorly understood since one gene can be implicated in different diseases (eg, LRRK2 mutations cause PD, LBD or PSP). (4) The specific impact of modifying factors, like low-risk mutations, lifestyle and exposures to insults on disease onset, initiation site and progression, is unknown at this time. (5) α-syn pathology can cause different pathologies and affect neuron or glia (eg, PD and MSA); thus, the factors regulating the vulnerability to α-syn neurotoxicity remain to be elucidated. A recent report suggests that different aggregated forms of α-syn (equivalent to prion strains??) lead to different pathologies when injected in rats,98 but the clinical relevance of this observation is unclear (different forms of PD; PD vs LBD??). In all, continuous genetic discoveries are contributing to further dissect the mechanisms of parkinsonism, but much work is still needed to understand the pathophysiology of PD and related disorders with sufficient detail to develop effective disease-modifying therapies, including the highly promising active and passive immunotherapy approaches.91 96 97
Acknowledgments
We are thankful to the many labs that have contributed to this field and apologise to those whose work we could not cite. The authors are thankful for institutional support for performing the literature research and writing the review.
References
Footnotes
Contributors PLZ and PFF conceived the study. YC, CHZ and YXW performed literature search and contributed to the draft. PLZ and PFF completed the draft. PFF created the figures. All authors read and approved the final version.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.