Article Text

Abstract
Background Until recently, determining penetrance required large observational cohort studies. Data from the Exome Aggregate Consortium (ExAC) allows a Bayesian approach to calculate penetrance, in that population frequencies of pathogenic germline variants should be inversely proportional to their penetrance for disease. We tested this hypothesis using data from two cohorts for succinate dehydrogenase subunits A, B and C (SDHA–C) genetic variants associated with hereditary pheochromocytoma/paraganglioma (PC/PGL).
Methods Two cohorts were 575 unrelated Australian subjects and 1240 unrelated UK subjects, respectively, with PC/PGL in whom genetic testing had been performed. Penetrance of pathogenic SDHA–C variants was calculated by comparing allelic frequencies in cases versus controls from ExAC (removing those variants contributed by The Cancer Genome Atlas).
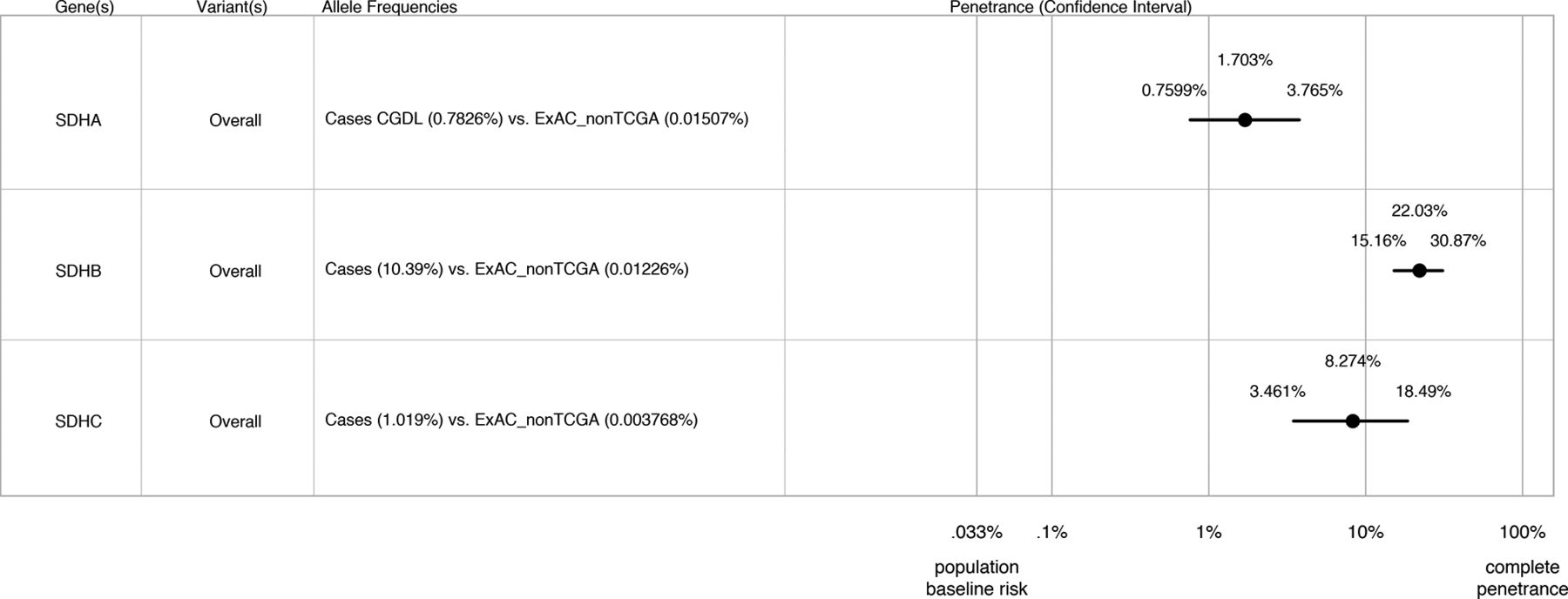
Results Pathogenic SDHA–C variants were identified in 106 subjects (18.4%) in cohort 1 and 317 subjects (25.6%) in cohort 2. Of 94 different pathogenic variants from both cohorts (seven in SDHA, 75 in SDHB and 12 in SDHC), 13 are reported in ExAC (two in SDHA, nine in SDHB and two in SDHC) accounting for 21% of subjects with SDHA–C variants. Combining data from both cohorts, estimated lifetime disease penetrance was 22.0% (95% CI 15.2% to 30.9%) for SDHB variants, 8.3% (95% CI 3.5% to 18.5%) for SDHC variants and 1.7% (95% CI 0.8% to 3.8%) for SDHA variants.
Conclusion Pathogenic variants in SDHB are more penetrant than those in SDHC and SDHA. Our findings have important implications for counselling and surveillance of subjects carrying these pathogenic variants.
- pheochromocytoma
- paraganglioma
- succinate dehydrogenase
- penetrance
- pathogenic variant
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Phaeochromocytomas (PCs, tumours of the adrenal medulla) and paragangliomas (PGLs, tumours of sympathetic or parasympathetic ganglia) are highly heritable, with 14 PC/PGL susceptibility genes identified.1–3 Six of these genes were included in the American College of Medical Genetics recommendations for mandated reporting of incidental findings from clinical exome and genome sequencing4 5: VHL, RET, succinate dehydrogenase subunits B, C, D (SDHB, SDHC, SDHD) and SDHAF2. The high heritability of PC/PGL strongly suggests that germline genetic testing be considered for all affected individuals, enabling predictive genetic testing for at-risk relatives if a pathogenic variant is detected.6 7
Germline mutations in the SDH genes are the most common genetic cause of PC/PGLs (MIM:168000,605373,115310 and 614165), occurring in approximately 15% of cases.1–3 By comparison, the next most common associated genes are VHL (4%–10%), RET (1%–5%) and NFI (1%–5%). VHL and NF1 are associated with Von Hippel Lindau (MIM:193300) and neurofibromatosis type 1 (MIM:162200), respectively, that include PC/PGL as part of a broader syndrome.
Since PC/PGL are rare tumours,8 not only should pathogenic variants be individually extremely rare but their cumulative frequency within disease-associated genes (if fully penetrant) should be <0.0001 in large population cohorts (ie, population prevalence of 1/5000 with allele frequency of 1/10 000 for autosomal dominant disease). Penetrance estimates from cohort studies varies considerably for each gene, ranging from ~3% for NF1 mutations to 90% for SDHD mutations.6 7 As a corollary of the observed imperfect penetrance, the frequency of potentially pathogenic variants within the population should be higher than the empiric figure presented above. It is also appreciated that individual pathogenic variants in the same gene may have differing functional impact and hence penetrance, a good example being the BRCA1 c.5096G>A (p.R1699Q) variant having moderate penetrance as opposed to the highly penetrant c.5095C>T (p.R1699W).9 This variable penetrance complicates genetic counselling.
Penetrance for SDHx variants has been somewhat controversial. Initial estimates suggested penetrance for SDHB variants of between 45% and 77% at ages 40–60 years,10–12 likely inflated, however, by inclusion of index cases. Subsequent analyses excluding index cases have suggested a much lower lifetime penetrance for SDHB variants of 22%–30%,11 13 14 although Rijken et al 15 have reported penetrance for SDHB variants of 42.1% (34.8%–49.5%) at 70 years and Jochmanova et al 16 reported penetrance of 49.80% (95% CI 29 to 74.9) at 85 years. Family-based penetrance studies in SDHB kindreds have suggested penetrance of 26%–35% by age 50 years.17–19 A large number of possible confounders might explain these differences, including referral bias, intensity of carrier screening, genotype–phenotype correlation or other genetic and/or environmental modifiers. A recent study of a relatively small number of SDHA variants reported penetrance of 39% at 40 years, but significantly less (13%) when index cases were removed.20 Penetrance for SDHC variants is as yet unknown.
An elegant approach to estimating penetrance of pathogenic variants was recently proposed by Vassos et al 21 and extended by Minikel et al 22 and Stessman et al,23 using an algorithm that compares variant allelic frequency in disease cases to its frequency in large population control cohorts such as the Exome Aggregate Consortium (ExAC), and accounting for known disease prevalence and proportion of hereditary cases for that disease.
In this study, we tested the hypothesis that allelic frequencies for pathogenic SDHA–C variants present in ExAC would be inversely proportional to their penetrance, using two cohorts of PC/PGL subjects in whom genetic testing had been performed. We excluded SDHD variants from this analysis since this gene is unsuited to Bayesian methodology due to imprinting and suboptimal coverage in whole exome sequencing.
Methods
Clinical methods
Australian patients with PC/PGL referred to the Cancer Genetics Diagnostic Laboratory, Royal North Shore Hospital, were tested for RET, VHL, SDHA, SDHB, SDHC and SDHD according to previously published methodology.24–26 Genetic testing was triaged initially by an in-house protocol and more recently according to PC/PGL Clinical Practice Guidelines,6 with the additional use of tumour SDHB immunohistochemistry27 to guide testing of SDH subunit genes. Testing of a sample was performed iteratively and stopped when a variant was identified and considered to be pathogenic/likely pathogenic (P/LP) by one of the following criteria: (1) the variant was described as P/LP in a disease-specific database (ARUP, Leiden Open Variation Database (LOVD) or ClinVar); (2) null variant or missense variant predicted to be damaging or deleterious by at least two in silico tools and a functional study to support damaging effect (eg, in the case of SDHx variants, loss of SDHB immunostaining in tumour) or (3) the variant was present in multiple affected family members. UK patients were analysed for SDHB/SDHC/SDHD/VHL mutations by Sanger sequencing (until 2012) and then mostly by a next generation sequencing assay of MAX, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127 or VHL.28 SDHB and SDHC sequence variants were classified as P/LP/benign/variants of uncertain significance by the reporting diagnostic laboratory. The GenBank Accession numbers were as follows: for SDHA NG_012339.1, NM_004168.3; for SDHB NG_012340.1, NM_003000.2 and for SDHC NG_012767.1, NM_003001.3.
Comparison of allele frequencies (table 1) between the Cohort Aus and Cohort UK was performed by G-test of Independence in DescTools R package V.0.99.24.
SDHA-C variants in cases from cohorts 1 (Aus) and 2 (UK) and also present in ExAC
Northern Sydney Local Health District Human Research Ethics Committee (Executive)noted that this project involves the use of existing data for the purpose of publishing figures on the occurrence of pathogenic variants. All subjects had given written informed consent for clinical genetic testing. The data being used are de-identified. Based on this information and in accordance with the National Health and Medical Research Council National Statement 2007—Section 5.1.22, the NSW Supplement to the National Statement—Section 5.1.6 and NSW Health Guideline GL2007_020: Quality Improvement and Ethics Review: A Practice Guide for NSW, this project was assessed as activity not requiring full HREC review.
LOVD search method
The LOVD (http://www.lovd.nl)29 was manually searched for variants in SDHA–C subunit genes and retrieved 59 unique variants in SDHA, 260 variants in SDHB and 66 variants in SDHC. Variants common both to LOVD and ExAC (http://exac.broadinstitute.org) and absent from The Cancer Genome Consortium (ftp://ftp.broadinstitute.org/pub/ExAC_release/release1/subsets/ExAC_nonTCGA.r1.sites.vep.vcf.gz) were tabulated according to allelic frequency in ExAC.
Penetrance and CI calculation
Bayesian calculation of the conditional probability of disease (penetrance) given the genotype was performed using the following formula23:

where
D
= disease,
G
= genotype, and  = absence of disease.
= absence of disease.
The denominator, equivalent to P(G), is the sum of joint probabilities of
G
with respect to both
D
and  which are mutually exclusive and collectively exhaustive of all possible events.
which are mutually exclusive and collectively exhaustive of all possible events.
 is the penetrance (the probability of disease given a genotype);
is the penetrance (the probability of disease given a genotype);  is the genotype frequency in cases;
is the genotype frequency in cases;  is the allele frequency in ExAC and
is the allele frequency in ExAC and 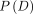 is the general population prevalence for PC/PGL, assumed ~1/3000.8
is the general population prevalence for PC/PGL, assumed ~1/3000.8
CI was obtained on the binomial probability as described by Rosenfeld et al.30 Upper bound CI for penetrance using upper bound on  and the lower bound on
and the lower bound on  . Lower bound CI for penetrance using lower bound on
. Lower bound CI for penetrance using lower bound on  and the upper bound on
and the upper bound on  . Data from cases and from ExAC were used to estimate these frequencies.
. Data from cases and from ExAC were used to estimate these frequencies.
Figures were plotted by using a modification of Minikel et al,22 and source code is provided online at https://github.com/ericminikel/prnp_penetrance.
Results
Cohort 1 consisted of 575 Australian subjects presenting with PC/PGL for whom genetic testing was performed between 1998 and 2016. Overall, 172 subjects (29.9%) with PC/PGL were diagnosed with a P/LP variant in one of nine genes. P/LP SDHA–C variants were identified in 106 subjects (nine SDHA, 90 SDHB and seven SDHC). By comparison, P/LP variants in other genes were: 36 SDHD, nine RET, 15 VHL, four TMEM127, one FH and one MAX.
Cohort 2 consisted of 1240 UK subjects presenting with PC/PGL for whom genetic testing was performed between 2001 and 2017.14 Overall, 446 subjects (36%) with PC/PGL were diagnosed with a P/LP variant in one of nine genes. P/LP SDHA–C variants were identified in 317 subjects (287 SDHB and 30 SDHC) and P/LP variants in other genes were 96 SDHD, 25 VHL, two RET, two FH, one TMEM127, two MAX and one SDHAF2.
We inferred pathogenicity for each variant from published evidence29 and/or based on segregation or loss of heterozygosity or the absence of protein on immunohistochemistry (table 1, online Supplementary table 1). Criteria for P/LP variants were consistent with standards for the interpretation of sequence variants issued by the American College of Medical Genetics and Genomics (ACMG).31
Supplemental material
We compared SDHA–C variants considered P/LP in either cohort against the high confidence variant calls in the ExAC database from which The Cancer Genome Atlas (TCGA) cases had been removed (obtained from ftp://ftp.broadinstitute.org/pub/ExAC_release/release1/subsets/, 22 June 2017) in order to diminish the risk of confounding by disease inclusion in cases. The allelic frequency of 13 variants that are present in ExAC are shown in table 1 and those that are not reported in ExAC are shown in the online Supplementary table 1. For completeness, we have listed in online Supplementary table 2 all previously reported SDHA–C variants from ClinVar that are also present in ExAC.
Supplemental material
For variants in SDHB and SDHC, allelic frequencies were not significantly different between Aus and UK cohorts: using G-test of independence, G=1.2858, 10 df, p=0.9995 and post hoc pairwise G-test found a coefficient value of 0.8897441 between the two cohorts. We therefore combined the two cohorts for subsequent analyses. We note that when all variants (including those not present in ExAC) were considered, SDHB variants were collectively more frequent in cohort 2 (23.2%) than in cohort 1 (15.6%) (table 1). This difference was not confined to a particular type of mutation (online Supplementary table 1) and therefore unlikely to be due to any systematic difference in variant detection method. The collective frequency of SDHC variants was similar in both cohorts (table 1).
Of these 13 P/LP SDHx variants in ExAC, all are individually rare with the exception of SDHA variant c.91C>T, p.Arg31* (frequency 1/3036). Although individually rare, when the population frequencies for these variants were combined together (excluding SDHA p.Arg31*), the estimated population prevalence of these hereditary PGL syndromes assuming complete penetrance would be 1/6000.
We next applied the principles described in Minikel et al 22, using the algorithm described by Stessman et al 23 and with CIs calculated as described by Rosenfeld et al 30 to estimate the lifetime penetrance of PC/PGL for SDHA–C variants taking into account allelic frequencies in our cases versus ExAC controls and estimated population prevalence of these disorders. Our penetrance estimates are shown in figure 1: predicted lifetime penetrance for SDHB variants is 22.0% (95% (CI 15.2% to 30.9%), for SDHC variants 8.3% (95% CI 3.5% to 18.5%) and for SDHA variants 1.7% (95% CI 0.8% to 3.8%). Penetrance estimates did not vary significantly by individual allele either in separate cohorts or in the combined analysis (online Supplementary figure 1). Although population stratification is not relevant when considering pathogenic variants causing monogenic disease (in the absence of a founder effect), nevertheless, to account for possible confounding by ethnicity, we also compared allelic frequencies in our cases against the European non-Finnish exome data from ExAC. As shown in the online Supplementary figure 2, exclusion of non-European/Finnish alleles from the control data set did not significantly alter our penetrance estimates although did result in wider CIs due to inclusion of fewer variants (for SDHA 1.2%, 95% CI 0.5% to 2.7%; for SDHB 20.1%, 95% CI 12.8% to 30.0% and for SDHC 10.4%, 95% CI 3.4% to 27.6%.
Supplemental material
Supplemental material
![[SP3.jpg]](https://jmg.bmj.com/content/jmedgenet/55/11/729/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
![[SP4.jpg]](https://jmg.bmj.com/content/jmedgenet/55/11/729/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
Estimated lifetime penetrance of pheochromocytoma/paraganglioma (PC/PGL) in subjects heterozygous for genetic variants in succinate dehydrogenase subunits A, B and C (SDHA, SDHB and SDHC) from cohorts 1 and 2 combined. The algorithm used to calculate these estimates is based on Minikel et al 22 and takes into account allelic frequencies in cases versus Exome Aggregate Consortium (ExAC) controls and estimated population prevalence of these disorders.
Discussion
We have systematically addressed the possibility of low penetrance alleles in hereditary PC/PGL syndromes, using a recently described approach for correlating penetrance with allelic frequency.22 Correct assignment of pathogenecity for genetic variants has become an urgent problem facing the clinical genetics community, particularly with increasing use of whole exome/genome sequencing technology.31 32 Our study has several notable results: first, P/LP SDHB, SDHC and SDHA variants are more common in ExAC than expected; second,our Bayesian estimate of lifetime penetrance for SDHB variants is close to empiric data from cohort studies and third, SDHC and SDHA variants have low penetrance.
Although each are individually rare, the collective frequency of known P/LP SDHB, SDHC and SDHA variants in ExAC was highly surprising and may have several explanations: (1) these hereditary endocrine disorders are more common than previously thought due to the presence either of subpenetrant alleles or incomplete case ascertainment; (2) development of these disorders requires additional genetic modifiers, the absence of which diminishes disease risk in carriers of P/LP alleles; (3) the ExAC database is inadvertently enriched for PC/PGL subjects (unlikely) or (4) that the ExAC database contains sequencing errors (unlikely). On one hand, it is attractive to dismiss these findings as variant calling artefacts present in the ExAC database; however, population frequency estimates for pathogenic BRCA mutations inferred in a similar fashion are extremely close to sequencing estimates from a randomly selected Australian patient pool in the Lifehouse study.33 Again, these estimates and population screening findings are at least twofold higher than previously perceived population estimates. That these variants are more common than expected is non-trivial, if whole exome/genome sequencing is performed at a population level, when apparently healthy subjects carrying so-called pathogenic alleles will outnumber subjects identified on the basis of disease expression34: if indeed present in 0.017% of the population, then ~4000 subjects in Australia and ~11 000 subjects in the UK are carrying these P/LP SDHx variants.
We deliberately excluded SDHD variants from our analysis, since this gene is unsuited to Bayesian methodology due to imprinting and suboptimal coverage in whole exome sequencing. (Only one of 37 different P/LP SDHD variants from our cohorts was present in ExAC, data not shown.) Paternally inherited SDHD variants are associated with high penetrance of disease35 and would therefore be expected to be rare in the general population.
The finding that SDHA c.91C>T, p.Arg31* occurs in ExAC at a population frequency >10−4 is at first glance surprising: several reports have shown an association between this variant with either PGLs or gastrointestinal stromal tumours36 37; it is more frequently reported in PGLs than expected by chance, and bona fide loss of function was inferred from tumoural loss of heterozygosity at this locus and by the absence of SDHA assessed by immunohistochemistry.37 However, familial disease appears to be rare in association with this variant20 consistent with low penetrance.
For SDHB variants, calculated lifetime penetrance estimates appear close to recent empiric data,11–14 and the lower penetrance estimates for SDHA and SDHC conform to our anecdotal experience. It is interesting to note that penetrance and risk of multifocal disease seem to be related, that is, ~30% subjects with SDHB variants will have more than one PGL or PC, whereas very few subjects with SDHA variants develop multifocal disease.20 This deserves further study with larger cohorts of specific genotypes.
Shah et al 38 recently used whole genome sequence data from 10 495 unrelated individuals (with replication in public data from more than 138 000 exomes/genomes in gnomAD) to study population frequency of pathogenic variants in ACMG-recommended 59 gene-condition sets, including SDHB and SDHD. They found that SDHB and SDHD P/LP variants were more than 10-fold inflated in the population compared with expected population prevalence of hereditary PC/PGL, and with one possible explanation being that some variants may have been misclassified. The alternate explanation that the inflation is due to incomplete penetrance is supported by our data with some frequent variants being significantly inflated in two clinically ascertained datasets in a consistent manner. Those 13 SDHx variants in ExAC that we have observed in our PC/PGL cases all have very strong evidence of pathogenicity in LOVD and/or ClinVar. Indeed, 12 of these variants have a ClinVar star rating of 2 (multiple submitters with assertion criteria). Moreover, nine of these variants are loss-of-function (premature termination or splice site) variants. That these variants are more frequent in the population than expected for the corresponding disease prevalence can only mean either that they are subpenetrant and/or that the disease itself is more common than realised. The fact that our Bayesian estimates for SDHB are so close to empiric findings from recent cohort studies11–14 and to family-based studies17–19 gives us confidence that our estimates for SDHC and SDHA are also reliable.
Our study has some important limitations. We deliberately chose a validation cohort from a population with close genetic similarity to the discovery cohort,39 and naturally our findings may not apply to populations with different ethnic backgrounds; indeed, it will be interesting to compare allelic frequencies of these variants in populations worldwide. These algorithms may underestimate penetrance for variants not present in ExAC; some studies12 14 15 have suggested that certain SDHB variants are more penetrant. With respect to using ExAC data as controls, we attempted to minimise confounding by using the data set from which TCGA cases had been removed; it is remotely possible that PC/PGL cases were inadvertently enriched in other cohorts contributing to ExAC (eg, within cardiovascular cohorts). Finally, it is possible that an iterative testing process may have missed combinations of pathogenic variants in two or more genes; although our subsequent experience using massively parallel sequencing approaches suggests that the presence of two germline pathogenic variants is rare (data not shown).
While our manuscript was under review, Maniam et al 40 reported a similar Bayesian approach to calculate penetrance for SDHA variants at 0.1%–4.9%, although their study was based on published series of SDHA cases rather than as we have done using PPGL case cohorts. Despite these differences in case ascertainment, the similarity of penetrance estimates between the two studies is striking and consistent with our conclusion that pathogenic SDHA variants are likely to have low penetrance for disease expression.
We conclude that this approach of using population frequency of suspected P/LP variants in ExAC is extremely useful to validate empiric calculations from cohort studies. Our data suggests that at least for P/LP variants present in ExAC, penetrance is approximately 22% for SDHB variants, 8.3% for SDHC variants and 1.7% for SDHA variants. Our findings will have critical value for genetic counselling and screening of subjects carrying these P/LP variants. By more robust stratification of risk, rational allocation of biochemical and imaging surveillance could reduce both the cost and anxiety associated with carrying a germline mutation.
Electronic database information
ExAC Browser, http://exac.broadinstitute.org.
Mutation Taster, http://www.mutationtaster.org/.
OMIM, http://www.omim.org/.
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/.
PROVEAN, http://provean.jcvi.org.
Acknowledgments
We are grateful to Dr Warren Kaplan and Dr Marcel Dinger for helpful discussions.
References
Footnotes
Contributors MF and RJC-B conceived the study and wrote the manuscript. DEB, TD, CL, ALR, BR and RJC-B were responsible for curating the genetic test results for Cohort 1, and KAA and ERM were responsible for Cohort 2. Additional oversight of the clinical cohorts was provided by JB, AC, AJG, RJH, HM, RS, LT, AT, and KT. DEB, MW and EK confirmed pathogenicity for each variant. DEB, YZ, MF and RJC-B performed the initial analyses, with input from ELD, RWT and AS. All authors had full access to the data, and contributed to review of the manuscript.
Funding This work was supported by NHMRC Project 1108032 to DEB, RT, ED, TD, KT, AJG, BGR, RH, AT and RJC-B and Hillcrest Foundation (Perpetual Trustees) to DB and TD.
Competing interests None declared.
Patient consent Not requried.
Ethics approval Northern Sydney Local Health District Human Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.