Article Text

Abstract
Non-epithelial ovarian tumours are rare neoplasms that occasionally arise in childhood and adolescence. They can be associated with various cancer susceptibility syndromes. The morphological overlap seen across these tumours and their rarity can make the diagnosis challenging. In the case of an incorrect diagnosis, the underlying genetic susceptibility may be missed. In this review, we outline the genetic background of ovarian non-epithelial tumours arising in children, emphasizing the genes harbouring pathogenic germline variants associated with each tumour type. Specifically, juvenile granulosa cell tumours, Sertoli-Leydig cell tumours, sex cord tumours with annular tubules, Sertoli cell tumours, germ cell tumours and small cell carcinoma of the ovary of hypercalcaemic type are discussed in this review. For each tumour type, we detail the personal and family history features and the presenting characteristics of the ovarian tumour as well as the pathological features and molecular markers that point towards a cancer predisposition syndrome. Throughout, we stress the need for specialised pathological review in difficult cases.
- ovarian tumour
- cancer predisposition syndrome
- cancer genetics
- pediatric oncology
Statistics from Altmetric.com
INTRODUCTION
Ovarian tumours in childhood and adolescence are uncommon and may be associated with cancer predisposition syndromes (CPSs). These tumours are distinguished from those that arise in adulthood by their histological subtypes and by the different CPSs with which they are associated. Clinicians should recognise that an ovarian tumour developing in childhood may be the first sign of a CPS in a family. Diagnosing the CPS offers the important possibility of surveillance for other CPS-related disease in the patient and family.
While many reviews outline the tumours associated with a CPS, very few describe the inverse relationship, that is, when should the clinician suspect a CPS following the diagnosis of an ovarian neoplasm. Here we discuss ovarian tumours that are most likely to arise in childhood and their associated CPSs. We also discuss clinical features that may indicate a higher likelihood of a CPS and cover current screening recommendations for these tumours. It is important to note that the most prevalent ovarian neoplasm associated with a CPS occurs in adults with predisposing BRCA1/BRCA2 mutations: high-grade serous carcinoma. However, these are epithelial neoplasms and are exceedingly rare in individuals under age 18 years. Mucinous epithelial neoplasms do occur under age 18 years but are not known to be associated with cancer susceptibility syndromes. Most paediatric ovarian tumours which are associated with a CPS are in the non-epithelial categories.
SEX CORD-STROMAL TUMOURS (SCST)
Juvenile granulosa cell tumour
General aspects
Juvenile granulosa cell tumours (JGCTs) (figure 1A) are rare ovarian tumours, accounting for less than 1% of ovarian neoplasms but accounting for approximately two-thirds of SCSTs presenting in childhood.1 2 Just under one-half of JGCTs are diagnosed in the first decade of life and an additional one-third in the second decade.1 Over 95% of patients present with disease limited to the ovary and these low-stage patients have an excellent prognosis, with >90% overall survival with surgical resection alone.3 Most tumours are unilateral, although 2%–5% of cases are bilateral.1 2 A common presenting feature of JGCTs is isosexual precocious pseudopuberty, as these tumours often secrete oestrogen.3 Rarely they are associated with androgenic manifestations.
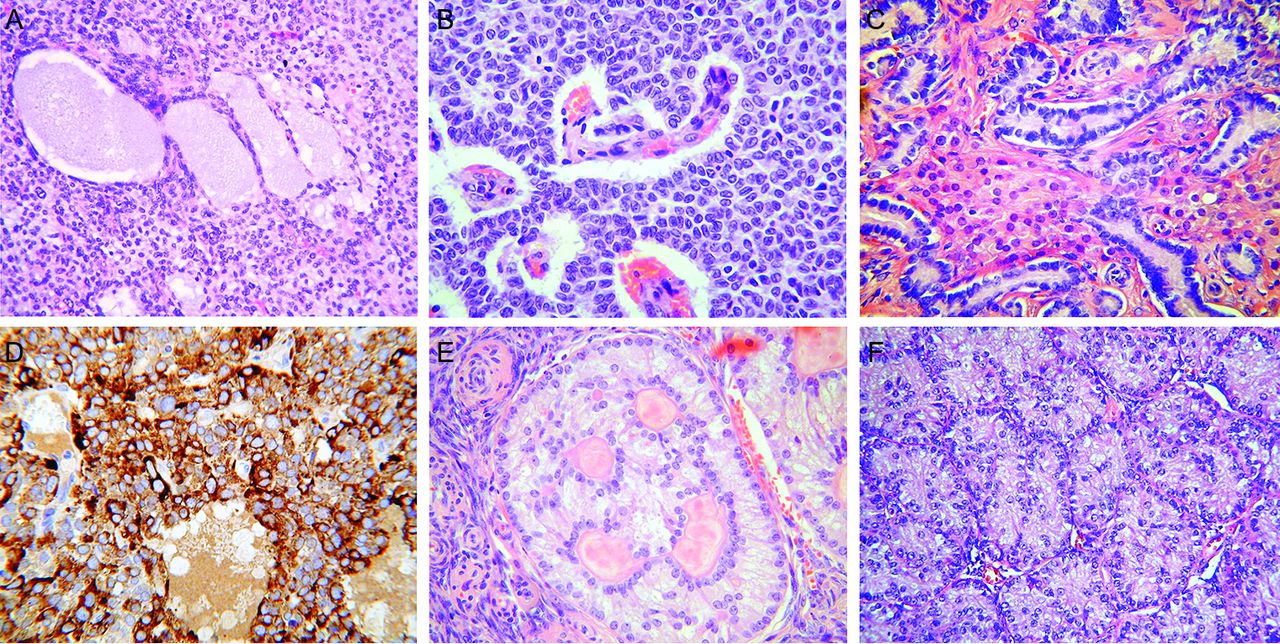
Various morphological types of ovarian SCST. These may exhibit considerable morphological overlap and expert pathology review may be required for a correct diagnosis. (A) Ovarian JGCT with diffuse arrangement of cells and larger follicle-like structures containing basophilic material. (B) Ovarian adult granulosa cell tumour composed of diffuse and microfollicular arrangements of tumour cells with bland vesicular nuclei. (C) Ovarian well differentiated Sertoli-Leydig cell tumour composed of Sertoli cell tubules with intervening Leydig cells with abundant eosinophilic cytoplasm. (D) JGCT which is diffusely positive with inhibin; this is a useful marker of ovarian SCSTs but does not allow distinction between the various types. (E) Ovarian sex cord tumour with annular tubules showing characteristic ‘punched-out’ spaces containing hyaline material. (F) Ovarian Sertoli cell tumour composed of solid Sertoli cell tubules. JGCT, juvenile granulosa cell tumour; SCST, sex cord-stromal tumour.
Key differential diagnoses
The differential diagnosis of JGCT includes other SCSTs and small cell carcinoma of the ovary of hypercalcaemic type (SCCOHT). The SCSTs which are most likely to be morphologically confused with JGCT are adult granulosa cell tumour (AGCT) (figure 1B) and Sertoli-Leydig cell tumour (SLCT) (figure 1C), although occasionally others may enter into the differential diagnosis. Immunohistochemistry (using markers such as inhibin, CD56, FOXL2, steroidogenic factor-1 and calretinin) is useful in diagnosing a SCST but not in distinguishing between the various types (figure 1D). From a molecular viewpoint, somatic hotspot missense FOXL2 mutations (c.402C>G; p.C134W) are found in approximately 95% of AGCTs, which are extremely rare in children, while the mutation is hardly ever reported in other SCSTs, including JGCT. Therefore, in cases where the differential includes AGCT and JGCT, demonstration of the presence or absence of a FOXL2 mutation may be extremely useful.4 5 Patients with SCCOHT may have hypercalcaemia and do not exhibit features of precocious pseudopuberty or other hormonal manifestations. Immunohistochemistry is useful in distinguishing between JGCT and SCCOHT since the former is usually inhibin positive and the latter negative. As discussed later, almost all SCCOHT lack expression of SMARCA4 (figure 2), while there is positive nuclear staining of JGCTs with this marker.

(A) Small-cell carcinoma of the ovary of hypercalcaemic type composed of small round blue cells with scant cytoplasm and with follicle-like formations. (B) There is loss of nuclear staining with SMARCA4 (BRG1) with a positive internal control in the form of staining of endothelial cells.
Cancer predisposition syndromes
JGCTs have been described in association with Ollier disease and Maffucci syndrome, subtypes of enchondromatosis syndromes (table 1). Tamimi and colleagues reported the first case of an ovarian JGCT in a 15-year-old girl with Ollier disease.6 Since then, 11 cases have been reported in the English literature.7 Ollier disease (OMIM #166000) and Maffucci syndrome (OMIM #614569) are skeletal disorders secondary to a dysplastic mesodermal process affecting the metaphyseal region of the short and long tubular bones. Although they are both types of non-spinal enchondromatosis, Maffucci syndrome includes soft tissue haemangiomas, while Ollier disease does not.8 They are considered non-hereditary disorders characterised by the presence of multiple enchondromas with asymmetric distribution with an estimated prevalence of 1/100 000.9 Somatic mutations in the IDH1 and IDH2 genes, classically affecting Arg132 of IDH1 or Arg172 of IDH2, have been identified in enchondromas and haemangiomas in patients with Ollier disease and Maffucci syndrome and have been linked to histone hypermethylation.8 10 A similar somatic mutation in these two genes has been suggested to explain the occurrence of JGCT, but this has yet to be confirmed.10 11
Features of paediatric ovarian tumours and their associated syndromes
Enchondromas that occur in Ollier disease and Maffucci syndrome become clinically apparent at a very young age and have the potential to transform into chondrosarcoma in up to 40% of patients.12 Following enchondromas, haemangiomas and chondrosarcoma, JGCT is considered the most common tumour in patients with Ollier disease or Maffucci syndrome. Most patients have had a diagnosis of enchondromatosis prior to the occurrence of the JGCT. Interestingly, many reports have described the ovarian tumour to be located ipsilaterally to the predominant skeletal dysplasia.13 As the long bones of the skeleton and the gonads are both derived from the mesoderm, some authors have hypothesised that involvement of the ovary is a manifestation of a generalised mesodermal dysplasia process.13
Other CPSs have occasionally been reported in patients with JGCT. Plon and colleagues described a child with a JGCT who had a germline mutation in PTEN and TP53.14 DICER1 somatic hotspot mutations have occasionally been identified in JGCTs, and two cases of JGCT have been associated with DICER1 syndrome,15 but a definitive link between JGCT and DICER1 mutations requires a larger series of cases. As DICER1 mutations are common in ovarian SLCT, it is possible that those JGCTs with the mutation represent misdiagnosed SLCTs; as discussed there may be morphological overlap between the two tumour types and JGCT-like areas may be seen in SLCTs.11 The study of additional cases of JGCT associated with DICER1 mutations, with expert pathological review to confirm the diagnosis, is required given these previous reports.15 Similar comments pertain to gynandroblastoma, a rare mixed ovarian SCST often exhibiting areas of both JGCT and SLCT; rarely these have been shown to contain DICER1 mutations and it is possible that some of these represent misdiagnosed SLCTs with JGCT-like areas.
As only a handful of cases of JGCT have been reported in the context of enchondromatosis syndromes and the link between other CPSs is not established, we have very little evidence on which to base genetic counselling recommendations. However, physical examination in patients with JGCT should include evaluation for features of enchondromatosis (table 1). Equally, there are no standardised management strategies for ovarian tumour surveillance in enchondromatosis syndromes. Although there are no consensus guidelines, we recommend that regular follow-up of growth and pubertal developmental be part of the surveillance for JGCTs in females with Ollier disease or Maffucci syndrome. As JGCTs are rare and characteristically benign, it seems reasonable not to incorporate specific ovarian imaging surveillance in enchondromatosis syndromes, unless clinically indicated.
Sertoli-Leydig cell tumours (SLCTs)
General aspects
SLCTs are an uncommon SCST, comprising 1%–2% of paediatric ovarian cancers.16 In a majority of cases, they are unilateral and confined to the ovary. They have been reported in females between the ages of 2 and 75, but most commonly occur in the second and third decades of life.17 SLCTs are composed of Sertoli and Leydig cells in varying proportions; according to the 2014 WHO Classification, they are categorised into well, moderately and poorly differentiated forms.18 Heterologous elements (including mucinous epithelium, rhabdomyoblastic and cartilaginous stromal elements, foci of carcinoid tumour) and/or a retiform pattern may be seen in moderately and poorly differentiated variants.18
Patients often present with hormonal manifestations, especially androgenic (leading to features of virilisation) and more uncommonly oestrogenic, including precocious puberty and menstrual abnormalities.17 Retiform tumours often develop at a younger age and are less associated with endocrine manifestations.19 The degree of differentiation of SLCTs has been shown to correlate with the patient’s prognosis, with well-differentiated forms almost always behaving in a benign fashion while poorly differentiated variants often exhibit malignant behaviour.17
Key differential diagnoses
SLCTs often result in diagnostic difficulties for pathologists in part because of their rarity and because they have a wide differential diagnosis (box). There may be considerable morphological overlap with other SCSTs and, as discussed, immunohistochemistry is of little or no value in distinguishing between the various SCSTs (figure 1D). However, immunohistochemistry is useful to distinguish SLCT from endometrioid carcinoma with which it may occasionally be confused. Similarly, retiform SLCTs may be confused morphologically with serous epithelial neoplasms but the latter occur at a much older age and the immunophenotype is different.
Cancer predisposition syndromes
SLCTs often manifest in the context of DICER1 syndrome (OMIM #601200), which is characterised by germline mutations in the DICER1 gene. In addition to SLCT, patients with this syndrome are at risk for developing very specific, typically rare paediatric neoplasms as well as more common neoplasms in adulthood (table 1, figure 3A).

{kind=link}
{kind=link}
{kind=link}
Pedigrees of families with manifestations of DICER1 syndrome and RTPS2. (A) Family with DICER1 syndrome-related manifestations. Affected relatives had a germline DICER1 variant: c.1525C>T (p.Arg509X). (B) Two sisters with SCCOHT who inherited a germline deletion of the entire SMARCA4 gene from their unaffected father. Black=affected, White=unaffected. +/+=wild type, +/–=heterozygous for pathogenic variant. MNG, multinodular goitre; RTPS2, rhabdoid predisposition syndrome type 2; SCCOHT, small cell carcinoma of the ovary of hypercalcaemic type; SLCT, Sertoli-Leydig cell tumour.
Patients with DICER1 syndrome typically have a loss of function germline mutation in DICER1 that can occur anywhere along the gene, along with a somatic missense DICER1 mutation in trans, most often in the RNAse IIIb ‘hotspot’ region.20 21 In non-hereditary cases of DICER1-related tumours, patients have two somatic mutations in DICER1 with no germline mutation. SLCTs are by far the most common gynaecological manifestation of the DICER1 syndrome and a recent study showed that 100% of moderately and poorly differentiated SLCTs contain either germline or somatic DICER1 mutations.22 Conversely, this same study suggested that well-differentiated SLCTs are not associated with germline or somatic DICER1 mutations.22 However, the number of well-differentiated SLCTs was small and currently we recommend that all patients with SLCT should be referred to a genetics service for consideration of DICER1 testing. This study also showed that moderately and poorly differentiated SLCTs without DICER1 mutations were misclassified neoplasms of some other tumour type, reinforcing the necessity for accurate pathology diagnosis (box).
In patients with SLCT, the presence of either bilateral tumours (a very rare phenomenon), a young age of onset or a personal or family history of a DICER1-related neoplasm should raise suspicion for a genetic cause.23 24 While the presence of a germline DICER1 mutation does not currently have an impact on treatment or survival, it is important for clinical follow-up of the patient as well as for surveillance and education for other at-risk family members. For affected individuals and parents of younger girls with DICER1 mutations, recognising the presenting clinical signs of SLCTs may be beneficial. Parents of patients with germline mutations may be unaffected due to either the low penetrance of DICER1 mutations in the parent or a de novo variant in the patient. For patients with DICER1-related ovarian tumours, consensus recommendations from the 2016 American Association for Cancer Research Childhood Cancer Predisposition Workshop have recently been published.25 For more information on DICER1 syndrome, visit http://www.dicer1syndrome.ca.
Additionally, SLCTs have been seen in young women and children with Peutz-Jeghers syndrome (PJS; see next section for details on PJS).26 27 However, a definitive link between SLCT and PJS requires further study.
Challenges in the pathological diagnosis of ovarian tumours in the context of CPS
An individual pathologist may never encounter certain cancer predisposition syndrome (CPS)-related ovarian neoplasms (most of which are non-epithelial) throughout their career due to their rarity.
Morphological overlap among ovarian sex cord-stromal tumours (SCSTs) may lead to ‘picture-matching’ and a potentially incorrect diagnosis. This can result in inappropriate management and a missed association with a CPS.
Expert pathology review and specific immunohistochemical markers or molecular testing may be required for correct diagnoses, especially in the paediatric population.
While immunohistochemical markers are useful in diagnosing an ovarian SCST, they are of little or no value in distinguishing between the morphological types.
When uncommon tumours are assigned to a CPS, journal editors and reviewers must ensure, as far as possible, that the pathological description is adequate and accurate.
Sex cord tumour with annular tubules (SCTAT)
General aspects
Ovarian SCTAT is a rare neoplasm, representing approximately 1%–2% of ovarian SCSTs.28 29 They have been reported in females aged between 5 and 57 years, with slightly less than half presenting at ≤18 years.30 31 Most tumours are confined to the ovary, are rarely associated with hormonal manifestations and follow a benign course.30 31 While most of the time SCTAT is not associated with a genetic syndrome, in 30%–35% of cases it occurs in the setting of PJS. The non-syndromic cases are usually unilateral, large and symptomatic. They may exhibit malignant behaviour with spread beyond the ovary. Those associated with PJS typically occur in younger patients (mean age 28 years versus 34 years in non-syndromic cases) and are often an incidental microscopic finding in ovaries removed in females known to have this syndrome.31 They are typically small, bilateral and multiple and clinically benign, although a small proportion exhibit malignant behaviour with extraovarian spread.
Key differential diagnoses
The morphological features of SCTAT are characteristic and the diagnosis is often straightforward (figure 1E), although given the rarity of the neoplasm, many pathologists will never encounter a case.31 The differential diagnosis chiefly includes other SCSTs, especially AGCT, SCT and SLCT. However, the rare gonadoblastoma, a mixed germ-cell and SCST may also enter into the differential diagnosis.31
Cancer predisposition syndromes
There is a well-recognised association between SCTAT and PJS.28 31 PJS is an autosomal dominant condition characterised by mucocutaneous melanocytic lesions, gastrointestinal hamartomatous polyps and the development of various benign and malignant neoplasms (table 1). The pigmented macules arise in the first few years of life in 90% of individuals; these may however fade with age. Approximately one-third of patients with PJS have symptoms related to gastrointestinal polyps by age 10 and two-thirds by age 20.32 The mean age of onset of ovarian tumours, particularly SCTAT (mean 28 years old), is lower than that of all other PJS-related cancers.33 Patients with PJS may also develop other gynaecological neoplasms such as other ovarian SCSTs, non-HPV-related cervical adenocarcinomas (gastric-type) and mucinous neoplasms at various sites such as the ovary. Males with PJS can also develop testicular large-cell calcifying Sertoli cell tumours.
A germline mutation in the tumour suppressor gene STK11 (also called LKB1) is the underlying cause of PJS in approximately 80%–90% of clinically affected families, without significant variation among racial and ethnic groups.34 Almost half of affected individuals have no family history compatible with PJS and the de novo mutation rate is estimated to be 25%, demonstrating the value of genetic testing for diagnosis.35 More than 300 causative variants in the STK11 gene have been reported, the majority of which are loss of function, although missense variants and partial or whole gene deletions have been described.36 Somatically, loss of heterozygosity at 19p13.1 is seen in the SCTATs of patients with PJS.37 A handful of studies have attempted to describe genotype-phenotype correlations with conflicting results.38–40
The precise lifetime risk for a female carrier of an STK11 germline mutation to develop a SCTAT is unknown but likely underestimated as they are often small and asymptomatic.41 The cumulative risk of developing an ovarian tumour in a STK11 germline mutation carrier is approximately 20% between the age of 15 and 64 years, with SCTAT accounting for a majority of the ovarian tumours.31 42
When a diagnosis of an ovarian SCTAT is made, clinicians should pay attention to aspects of the personal history, family history and tumour characteristics, which could suggest the presence of PJS (table 1). Considering the rarity of this ovarian tumour and its specific association with PJS, we recommend sending all children with SCTAT for a formal genetic evaluation and testing for a germline STK11 mutation. Currently, no specific recommendations exist for surveillance for gonadal tumours in children with PJS. One group suggests starting yearly surveillance at age 25 with pelvic examination, pap smear and transvaginal ultrasound.33 This surveillance is not specific to SCTAT, but is used for all gynaecological cancers. Nevertheless, a female carrier of PJS should be examined at least yearly with special attention paid to symptoms and signs which may suggest hormonal changes.43
Sertoli cell tumours
General aspects
Sertoli cell tumours account for <5% of ovarian SCSTs and predominantly present in women of reproductive age (mean 30 years).44 In a series of 54 patients (age range 2–76 years) reported by Oliva and colleagues, 14% of patients were prepubertal.44 These tumours commonly result in hormonal manifestations, especially oestrogenic and less commonly androgenic or progestogenic.19 44 Sertoli cell tumours (figure 1F are most often benign neoplasms confined to the ovary and have an excellent prognosis with low reported rates of recurrence.45 Rare lipid-rich and oxyphilic subtypes of Sertoli cell tumour have been reported.45 46
Key differential diagnoses
Ovarian Sertoli cell tumours may have a wide differential diagnosis which can include other SCSTs (SLCT, AGCT, SCTATs), endometrioid carcinoma, carcinoid tumour and various metastatic adenocarcinomas.44 A correct diagnosis and distinction from the other neoplasms in the differential may be facilitated by a panel of immunohistochemical markers.
Cancer predisposition syndromes
Sertoli cell tumours, particularly the lipid-rich and oxyphilic subtypes, have been described in children and young women with PJS.47 In the series of Oliva and colleagues, 6/54 patients with Sertoli cell tumours were reported to have PJS, of which lipid-rich and oxyphilic subtypes were predominant.44 In another report of 16 SCSTs from patients with PJS, four were lipid-rich and two were oxyphilic Sertoli cell tumours, with most of these SCSTs arising in the first two decades of life (range 4–30 years).27 Conversely, in a study by Tavassoli and Norris of 28 patients with ovarian Sertoli cell tumour (age range 7–79 years), four had lipid-rich variants and none had signs of PJS.45 In males with PJS, oestrogen-producing testicular Sertoli cell tumours have been reported.48
When a diagnosis of a Sertoli cell tumour is made, clinicians should be aware of certain aspects of the personal history, family history and tumour characteristics (such as lipid-rich or oxyphilic subtypes) that raise the possibility of PJS (table 1). It seems reasonable to send a child with an ovarian Sertoli cell tumour for genetic counselling and testing for STK11 mutations only if the clinician has a higher suspicion based on additional features.
GERM CELL TUMOURS
General aspects
Malignant germ cell tumours comprise 75% of ovarian neoplasms arising in the first 20 years of life and account for 15% of malignancies in adolescents aged 15–19 years old.47
Ovarian germ cell tumours are a heterogeneous group of neoplasms arising from pluripotent primordial germ cells, including teratoma (mature and immature), choriocarcinoma, yolk sac tumour (YST; also called endodermal sinus tumour), dysgerminoma, embryonal carcinoma and mixed subtypes. Teratomas (the majority of which are mature) are the most common ovarian germ cell tumours, followed by dysgerminoma and YST.49 Gonadoblastoma is a mixed germ cell-SCST with Y-chromosome material that characteristically develops in the context of gonadal dysgenesis. Although considered benign, gonadoblastomas have the potential to evolve into a dysgerminoma and exhibit malignant behaviour.50 51
Ovarian germ cell tumours most frequently present with a palpable mass and abdominal pain, and approximately 10% can present with an acute abdomen as a result of ovarian torsion, haemorrhage or rupture.52 Certain subtypes may be associated with an elevated serum βHCG or alpha-fetoprotein (αFP).53 Malignant germ cell tumours are highly curable, with 89%–98% survival rates.49 Adverse prognostic factors include advanced tumour stage and elevated serum markers.54 55
Key differential diagnoses
The differential diagnosis depends on the morphological subtype of germ cell tumour. Mature teratomas (dermoid cysts) are usually easily diagnosed. The differential diagnosis of some of the other morphological subtypes mayinclude a wide range of non-germ cell neoplasms. A variety of immunohistochemical markers, including αFP, placental alkaline phosphatase, OCT3/4 and SALL4, may assist in diagnosis.
Cancer predisposition syndromes
Increasing evidence supports a genetic predisposition to the development of germ cell tumours. Genetic syndromes and multiple sex-chromosome aneuploidy syndromes leading to various degrees of gonadal dysgenesis have been consistently associated with germ cell tumour development, particularly but not limited to gonadoblastoma.
WT1-related disorders, most commonly Frasier syndrome and Denys-Drash syndrome (DDS), are described in association with ovarian gonadoblastoma. Both diseases are associated with steroid-resistant progressive nephropathy, gonadal tumours and male pseudohermaphrodism and therefore may easily be missed when the patient is phenotypically female.56 The WT1 gene is a tumour suppressor gene that encodes a zinc finger transcription factor involved in regulation of early gonadal and kidney development.57 58 It consists of 10 exons, with two regions of alternative splicing in exons 5 and 9. The final nucleotide of exon 9 codes for lysine, threonine and serine (KTS) and a correct ratio of KTS is necessary for the normal function of this gene.
Frasier syndrome is typically caused by a point mutation on the donor splice-site mutation in WT1 on intron 9, leading to a decrease in +KTS isoforms.59 It results in a slowly progressive glomerulopathy leading to nephrotic syndrome often in the first decade of life and typically resulting in renal failure in the second or third decade.60 61 These patients have gonadal dysgenesis with streak gonads, which are associated with a high risk of gonadoblastoma. The frequency of unilateral or bilateral gonadoblastoma in these patients is reported to be between 37% and 60%.60 62 63 Patients with Frasier syndrome who developed dysgerminoma have been described, some of these tumours having a gonadoblastoma component.63 64 Gonadal tumours are reported in 67% of type 1 (female XY) and 37.5% of type 2 (male XY), but not in type 3 (female XX) Frasier syndrome.60
DDS results from a germline missense mutation in WT1 exons 8 or 9 that code for zinc fingers 2 or 3, leading to abnormal production of the WT1 protein.65 Most cases of DDS are caused by one of two missense mutations located in exon 9: c.1180C>T (p.Arg394Trp) or c.1186G>A (p.Asp396Asn).65 66 This results in early-onset nephrotic syndrome andprogression to renal failure, male pseudohermaphrodism and development of Wilms tumour.56 Wilms tumours are the most frequent malignancies seen in patients with DDS. Unilateral or bilateral gonadoblastomas are described in patients with DDS, classically presenting in children between age 1.3 and 2.6 years.67 In a series of 150 patients with DDS, the frequency of gonadoblastoma was reported to be 4%.56 Gonadoblastoma, Wilms tumour and renal parenchymal disease tend to develop early, with an average age of presentation of 1.7 years for tumours and 1.4 years for the renal manifestations.56
WAGR syndrome (Wilms tumour, Aniridia, Genitourinary anomalies and intellectual disability), caused by a partial deletion of chromosome 11p13, which includes the PAX6 and WT1 genes, has also been reported in cases of childhood ovarian gonadoblastoma, although to a lesser extent than Frasier syndrome or DDS.68
Gonadoblastoma has been reported in 10%–35% of patients with Turner syndrome with Y-chromosome material (45,X0/46,XY mosaicism).51 69 70 In a cohort of females with Turner syndrome, under 1% developed gonadoblastoma (only those with Y-chromosome material). This resulted in a cumulative risk of gonadoblastoma by age 25 years of 8%, which is likely an underestimate.71 Gonadoblastoma is seen in Turner syndrome and in the wide spectrum of other sex-chromosome aneuploidy syndromes associated to gonadal dysgenesis.
Following a diagnosis of an ovarian gonadoblastoma, a personal history of Wilms tumour, renal parenchymal disease, aniridia or an underlying urogenital malformation is highly suspicious for a WT1-related disorder (table 1). Given the rarity of familial WT1 disorders, family history may not be the most helpful indicator.72 Short stature or delayed puberty in a female with a gonadoblastoma should also trigger investigations for a sex-chromosome aneuploidy syndrome, specifically Turner syndrome. The latest guideline for intersex disorders recommends gonadectomy in phenotypic females with XY karyotype who have DDS and Frasier syndromes.73 74 However, Ezaki and colleagues suggest that gonadectomy could be avoided in patients with Frasier syndrome type 3.60 Gonadectomy is also recommended in Turner syndrome if Y-chromosome material is present.73 74
For ovarian germ cell tumours other than gonadoblastoma, the association with CPSs is not as clear. Familial cases of ovarian germ cell tumours and familial clustering of benign teratomas and ovarian dermoid cysts have also been described, although no genes have yet been implicated.75 76 Paediatric case reports of ovarian germ cell tumours in the context of various syndromes have been published (table 2).77–84 Of note, reports describing ovarian germ cell tumours in patients with ataxia-telangiectasia (A-T) have been published.85 86 A-T, caused by biallelic mutations in the ATM gene, is associated with progressive cerebellar ataxia, oculocutaneous telangiectasia, immunodeficiency and cancer risk, especially haematological malignancies. Pathogenic variants in ATM result in DNA repair defects, making it essential to identify this syndrome, as patients can develop excessive toxicity to cancer therapy.
Other CPS (non-sex chromosomal aneuploidy syndromes) associated with paediatric ovarian GCTs
Small cell carcinoma of the ovary of hypercalcaemic type
General aspects
SCCOHT is an uncommon, but well-known, ovarian neoplasm, the histogenesis of which has been unknown until recently; it is included in the category of miscellaneous ovarian neoplasms in the 2014 WHO Classification. While uncommon, SCCOHT is the most frequent undifferentiated ovarian cancer presenting in women below the age of 40, with a mean age at diagnosis of 24 years.87 It has been reported in females as young as 14 months,88 89 but most often arises in the second or third decade of life.87 In two-thirds of cases, the patients have serum hypercalcaemia. SCCOHT is an extremely aggressive cancer, with 65% of cases recurring following primary therapy and long-term survival rates of 33%–55% for early stage and 0%–30% for advanced stage neoplasms.90 91 Due to the aggressive nature of the tumour, a correct diagnosis at presentation is crucial.
Key differential diagnoses
Establishing a diagnosis of SCCOHT may be difficult, as there is a broad differential diagnosis. The differential diagnosis may include a wide range of neoplasms, including AGCT, JGCT, germ cell tumours, endometrial stromal sarcoma, malignant melanoma, primary and metastatic small cell neuroendocrine carcinoma and a variety of small round blue cell tumours, which highlight the importance of expert pathology review for accurate diagnosis (box).87 92 While the tumour cells in SCCOHT usually have scant cytoplasm resulting in a small cell appearance, some cases contain tumour cells with abundant eosinophilic cytoplasm, often resulting in a large cell appearance, sometimes with a rhabdoid morphology. As discussed below, SCCOHT is caused by mutations in the chromatin remodelling gene SMARCA4.90 These mutations are accompanied by loss of expression of the SMARCA4 (BRG1) protein in 97% of cases, and immunostaining for SMARCA4 has now been established as an extremely useful way to diagnose SCCOHT and distinguishes this entity from its mimics which almost always exhibit retention of nuclear immunoreactivity (figure 2).93
Cancer predisposition syndromes
It has recently been established that SCCOHT is a monogenic disease and caused primarily by germline and somatic deleterious mutations in the chromatin remodelling gene SMARCA4, a member of the SWI/SNF chromatin remodelling complex.90
Prior to the discovery of the association between SMARCA4 and SCCOHT, it was found that germline mutations in SMARCA4 can predispose patients to the development of rhabdoid tumours, including atypical teratoid/rhabdoid tumours, which are most often caused by mutations in another chromatin remodelling gene, SMARCB1.94 95 Germline mutations in SMARCB1 and SMARCA4 are associated with rhabdoid tumour predisposition syndrome types 1 and 2, respectively (RTPS1 and RTPS2); mutations were discovered to underlie the development of SCCOHT, and it was also recognised that these ovarian tumours are in fact morphologically very similar to rhabdoid tumours. Their similarity has led to the suggestion that SCCOHT is part of the rhabdoid tumour spectrum, and geneticists should now recognise that female carriers of pathogenic SMARCA4 mutations are at risk for both rhabdoid tumours and SCCOHT.
While RTPS2-associated SMARCA4 mutations are largely loss of function variants, a handful of missense variants have been described.90 These variants have been found to occur across the length of the gene, with no predominant hotspot regions.90 Missense variants in SMARCA4 and other members of the SWI/SNF complex more often cause Coffin-Siris syndrome, a developmental disorder not associated with tumour development, rather than RTPS2.96 Additionally, one case has been reported of a patient with a de novo SMARCA4 nonsense variant and concomitant Coffin-Siris syndrome, microphthalmia and SCCOHT.97
Although few familial cases have been described, studies have shown that up to 43% of patients with SCCOHT harbor a germline mutation in SMARCA4.90 This often occurs in patients with no family history of SCCOHT or rhabdoid tumours, and in these cases, the pathogenic mutation is usually inherited from the patient’s father.98 99 Penetrance of these mutations is still unknown.
Given the high incidence of SMARCA4 germline mutations in patients with SCCOHT, we recommend that once the diagnosis is established, all patients should be referred to a genetics service and undergo genetic counselling. Patients diagnosed below the age of 20 years are highly likely to carry a germline mutation.90 If a pathogenic mutation is found, all relevant relatives should undergo genetic counselling and testing for the familial variant. However, the unknown penetrance of these mutations and the young age of onset of this tumour makes it difficult to counsel mutation carriers and their families. Female carriers of truncating mutations are at risk for SCCOHT, and infant carriers of both genders may be at risk for rhabdoid tumours (table 1). As SMARCA4-mutated rhabdoid tumours have not been reported in patients older than 46 months, the development of these tumours in older carriers is unlikely.100 However, the oldest woman to date diagnosed definitively with SCCOHT (showing loss of SMARCA4 staining in her tumour) was 56 years old at diagnosis.90 Further testing of affected and unaffected family members will hopefully elucidate the true penetrance and allow carriers to be more informed when making potentially life-altering decisions.
Currently there are no established preventive recommendations for female carriers. Bilateral oophorectomy would likely be an effective preventive measure,101 but without penetrance data, determining the optimal age to consider this option is difficult. Prophylactic oophorectomy may be considered by young females known to carry a germline SMARCA4 mutation and has been performed in at least one adolescent carrier.99 The efficacy of ovarian surveillance for female carriers is still undetermined, but until data are available, ovarian imaging is a reasonable option.
Although the familial incidence of SCCOHT, and RTPS2 in general, is low, it is important to note the high fraction caused by germline SMARCA4 mutations and to recognise that even without a family history, the tumour may be hereditary if the patient has inherited a germline mutation from her father or acquired one de novo (figure 3B). Furthermore, it is important to note that infant carriers of both genders may be at risk for rhabdoid tumours in addition to SCCOHT.
CONCLUSION
Ovarian tumours in the paediatric population are not common, but in cases where they arise, consideration should be given to a possible underlying CPS, especially with a SCST. A thorough personal and family history should be collected as well as details on the tumour that may increase its likelihood of being the result of a germline mutation in a cancer predisposition gene. Although we have highlighted the most commonly known genes associated with these syndromes, novel sequencing techniques may uncover new genes and syndromes associated with uncommon ovarian tumours. Discovery of new gene-disease associations is outpacing clinical guidelines and physicians should always remain suspicious of a CPS following the diagnosis of an uncommon/rare ovarian tumour in a paediatric patient.
Acknowledgments
The authors would like to thank Dr John R Priest for his assistance with this manuscript and the identification of one of the families illustrated in this review. We also thank Marie Jeanjean for her assistance with one the families presented in this review.
References
Footnotes
CG and LW are the co-first authors.
Contributors CG, LW and SV: implicated in all aspects of this review and cowrote the manuscript;. WGMC: creation of the figures, revision of the manuscript draft;. WDF: review conception and revision of manuscript draft.
Funding WDF acknowledges the funding support of the Canadian Institutes of Health Research (FDN: 148390). CG’s research was generously funded by a grant from the Cedars Cancer Foundation and the Montreal Children’s Hospital Foundation.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.