Article Text

Abstract
Background Hydranencephaly is a congenital anomaly leading to replacement of the cerebral hemispheres with a fluid-filled cyst. The goals of this work are to describe a novel autosomal-recessive syndrome that includes hydranencephaly (multinucleated neurons, anhydramnios, renal dysplasia, cerebellar hypoplasia and hydranencephaly (MARCH)); to identify its genetic cause(s) and to provide functional insight into pathomechanism.
Methods We used homozygosity mapping and exome sequencing to identify recessive mutations in a single family with three affected fetuses. Immunohistochemistry, RT-PCR and imaging in cell lines, and zebrafish models, were used to explore the function of the gene and the effect of the mutation.
Results We identified a homozygous nonsense mutation in CEP55 segregating with MARCH. Testing the effect of this allele on patient-derived cells indicated both a reduction of the overall CEP55 message and the production of a message that likely gives rise to a truncated protein. Suppression or ablation of cep55l in zebrafish embryos recapitulated key features of MARCH, most notably renal dysplasia, cerebellar hypoplasia and craniofacial abnormalities. These phenotypes could be rescued by full-length but not truncated human CEP55 message. Finally, we expressed the truncated form of CEP55 in human cells, where we observed a failure of truncated protein to localise to the midbody, leading to abscission failure and multinucleated daughter cells.
Conclusions CEP55 loss of function mutations likely underlie MARCH, a novel multiple congenital anomaly syndrome. This association expands the involvement of centrosomal proteins in human genetic disorders by highlighting a role in midbody function.
- hydranencephaly
- midbody
- multinucleated neurons
- Potter sequence
- renal dysplasia
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Hydranencephaly is a congenital anomaly with a frequency of ∼1 in 10 000 births.1 It is defined as the almost complete absence of the cerebral hemispheres, replacement of the missing neural tissue with fluid-filled sacs, relative preservation of the posterior fossa structures and a normal intracranial compartment. On prenatal ultrasonography, hydranencephaly may resemble extreme hydrocephalus, but can be differentiated through fetal or postnatal MRI. It is usually an isolated anomaly, although there are reports of syndromic associations. Underlying causes are thought to include teratogens (eg, congenital infections, valproic acid exposure), chromosomal disorders (eg, trisomy 13, triploidy) and single-gene disorders (eg, proliferative vasculopathy, X-linked hydranencephaly with abnormal genitalia).2 The pathophysiology in the majority of cases of hydranencephaly is thought to be early intracranial vascular obstruction in a fetus, leading to necrosis and cystic degeneration of the cerebral hemispheres, although there may be other mechanisms.2–4
Here we report a family with an apparently autosomal recessive form of hydranencephaly that includes additional congenital anomalies and dysmorphisms. Literature review shows that this is a rare, but consistent, syndromic association and provides additional evidence for a recessive inheritance pattern. Neuropathological analysis identified a unique pathophysiology underlying the hydranencephaly likely related to arrested neuronal mitosis, suggesting that this anomaly is probably caused by non-vascular mechanisms. Through the combined application of homozygosity mapping and exome sequencing, we identified the likely causative genetic defect, a nonsense mutation in CEP55 (c.1274C>A; p.S425X), which encodes centrosomal protein-55 kDa (CEP55). Although not implicated previously in any genetic disorder, CEP55 is known to be essential for the final step of cytokinesis, abscission and has also been found to be upregulated in some tumours.5 More recently, loss of cep55l, the zebrafish ortholog of CEP55, was also shown to result in various developmental defects in zebrafish embryos.6 In the present study, we link aberrant fetal development in humans with a CEP55 mutation; we reproduce several of the key phenotypes in zebrafish models and we show that the observed C-terminal truncation of the protein likely interferes with the midbody functions of CEP55, thereby providing insight into pathomechanism.
Methods
Patient material
The study was conducted on a single Canadian family of Dutch-German Mennonite ancestry. Postmortem sampling, pathological analysis, karyotype and comparative genomic hybridisation were done during the provision of clinical care using standard protocols. All subjects (or legal guardians) gave written informed consent to participate.
Genetic mapping
Genotyping of the nuclear family was performed using Illumina Human Omni 2.5 SNP arrays at the Centre for Applied Genomics, Toronto, Canada. These data were analysed using the AutoSNPa and Phaser SNP mapping tools developed at the University of Leeds.7 ,8 Additionally, X-linked markers were sorted manually in a spreadsheet to determine X-chromosome segregation.
Exome sequencing
Sequencing and analysis were conducted as described.9 Briefly, we enriched for the coding sequence regions with the Agilent SureSelect 50 Mb (V3) All Exon Kit (Agilent Technologies, Santa Clara, California, USA). The resultant library was sequenced with an Illumina HiSeq platform to generate >14 Gb of 100 bp paired-end reads. Read alignment to the reference genome (hg19) was done with the Burrows-Wheeler Aligner. Picard and ANNOVAR were used to identify and annotate variants. Variants of low quality that did not meet objective criteria (read depth (DP)>5; quality (Q) ≥50; mapping quality (MQ)≥4; quality by depth (QD)<1.5) were excluded. Coverage of Consensus Coding Sequence (CCDS) bases was assessed using the Genome Analysis Toolkit. We assessed population frequency of variants using dbSNP132, the 1000 Genomes data set (2012/04 release) and the 6500 exomes in the NHLBI Exome Variant Server. In addition, we assessed variant frequency using the Genome Quebec Innovation Centre database (∼800 exomes). We confirmed all candidate variants using Sanger sequencing.
RNA isolation from patient cells and expression studies using RT-PCR
We isolated total RNA from fibroblasts derived from a healthy individual (3384) and a multinucleated neurons, anhydramnios, renal dysplasia, cerebellar hypoplasia, and hydranencephaly (MARCH) patient using the RNeasy Mini Kit (QIAGEN) according to the manufacturer's instructions. cDNA was made using SuperScript III First-Strand Synthesis System and oligo(dT)20 primers (Life Technologies) according to manufacturer's instructions. We PCR amplified cDNA using a Veriti 96-well Thermal Cycler (model 9902, Applied Biosystems) and the following conditions: 95°C for 5 min, followed by 27 cycles of 95°C, 60°C and 72°C of 30 s periods each and 7 min of 72°C. Specifically, we amplified CEP55 exons 7–9 (390 and 262 bp) and exons 2–3 (287 bp), respectively, and a GAPDH control fragment (288 bp). We obtained primers from Sigma-Aldrich. The CEP55 390 bp fragment I was amplified with forward and reverse (5′-3′) primers ‘CCAGTTTCTTTACACATCTCTGC’ and ‘CAGTATTCCACATGGACAAGC’, the 262 bp fragment II with ‘CCAGTTTCTTTACACATCTCTGC’ and ‘TCTGTTTTCAGTCTCTCCTTGG’, and the 287 bp fragment III with ‘AAGTAAGTGGGGATCGAAGC’ and ‘TCTCTTCCAGCTGTTCAAGC’. The 5′-3′ sequences of the GAPDH primers are ‘TCTCCTCTGACTTCAACAGCGA’ and ‘TACATGGCAACTGTGAGGAGGG’. The CEP55 390 bp fragment was Sanger sequenced to validate the absence or presence of the c.1274C>A (p.S425X) mutation in control and patient cDNA, respectively.
Transient gene suppression in zebrafish embryos
All zebrafish studies were conducted with approval from the Duke University Institutional Animal Care and Use Committee. Two splice-blocking morpholino (MO) antisense oligonucleotides targeting cep55l (sb1, 5′-CTGAGATAAAGCGACTAATTCACCT-3′ and sb2, 5′-GTTGAGCTTTTCTTACCATCAGACT-3′) were obtained from Gene Tools, LLC. To determine the optimal dose for in vivo complementation assays, we injected 4, 5 and 6 ng of MO into one-to-four-cell stage zebrafish embryos obtained from natural matings of heterozygous -1.4col1a1:egfp transgenic adults.10 To determine MO efficiency, we harvested embryos in Trizol (Invitrogen), extracted total RNA according to the manufacturer's instructions and generated oligo-dT-primed cDNA with the QuantiTect reverse transcription kit (Qiagen) for use as RT-PCR template. Sanger sequencing of RT-PCR products was used to identify the precise alteration of endogenous transcript. For rescue experiments, 5 ng MO and 100 pg RNA were used either independently, or combined.
CRISPR/CAS9 genome editing of cep55l
We used the CHOPCHOP design tool to identify cep55l guide (g)RNA target sequences, for which one pair of oligonucleotides were synthesised and annealed.11 Annealed oligos were ligated into the T7cas9sgRNA2 vector as described,12 and plasmids were sequence confirmed. Template vector was linearised with BamHI and gRNA was transcribed with the MEGAshortscript T7 kit (Life Technologies). In total, 100 pg of cep55l gRNA and 100 pg of CAS9 protein (PNA Bio) was co-injected into the cell of one-cell stage embryos. To determine targeting efficiency in F0 mutants, we harvested single embryos at 2 days post-fertilisation (dpf) and 4 dpf, extracted genomic DNA and PCR amplified a region flanking the gRNA target site. To estimate the percent mosaicism, PCR products were cloned into a TOPO-Therapeutic abortion (TA) vector (ThermoFisher); plasmid was prepped from multiple individual colonies (n=19–54) per embryo (n=5–8 embryos/condition) and Sanger sequenced on an ABI3730.
NGS library preparation and sequencing for CRISPR/CAS9 outcomes
Gene-specific primers were used to amplify a region flanking the gRNA target site; with tags 5′-CGCTCTTCCGATCTCTG-3′ and 5′-TGCTCTTCCGATCTGAC-3′ appended to the 5′ side of the gene-specific portion for forward and reverse primers, respectively. These amplicons were used as a template for the second PCR to add the Ion Xpress barcode adapters. Equimolar concentrations from each amplicon were pooled to make the final sequencing library. The library was diluted to 100 pM and sequencing template was prepared using the Ion PGM Hi-Q OT2 Kit on the Ion One Touch 2 instrument according to manufacturer's instructions. Templated Ion Sphere Particles were enriched on an Ion One Touch ES and sequencing was performed on an Ion Torrent PGM with the Ion PGM Hi-Q Sequencing Kit. Demultiplexing of barcoded samples was performed using Torrent Suite Software 5.0.4 and reads were trimmed using Cutadapt.13 CRISPR-Cas9 outcomes were quantified using CRISPResso computational pipeline.14
Automated zebrafish imaging
Larvae were positioned and imaged live with the Vertebrate Automated Screening Technology (VAST) platform (Union Biometrica) as described.15 Larvae were anaesthetised with 0.2 mg/mL Tricaine prior to being loaded into the sample reservoir. Dorsal and lateral images were acquired at a >70% minimum similarity from the pattern-recognition algorithm. Once recognised inside the 600 μm capillary of the VAST module on the microscope stage, larvae were rotated 180° to capture a fluorescent image of the jaw. NIH ImageJ software was used to measure the angle of the ceratohyal cartilage.
Whole mount TUNEL and immunostaining
We used the ApopTag rhodamine in situ Apoptosis Detection kit (Chemicon) as described.16 ,17 For whole-mount immunostaining, embryos were fixed in Dent's solution and stained with primary antibody (anti-phospho-histone H3 (ser10)-R antibody (sc-8656-R, Santa Cruz, 1:500); anti-acetylated tubulin (T7451, Sigma-Aldrich, 1:1000) and anti-NaK ATPase (a6F, DSHB;1:500)); and secondary detection was accomplished with Alexa Fluor goat anti-mouse IgG (A21207, Invitrogen; 1:1000) in blocking solution as described.16 ,18 ,19 Fluorescent signals were imaged on laterally or ventrally positioned larvae and z-stacked on a Nikon AZ100 microscope using NIS Elements AR software. We quantified staining by counting positive cells in defined regions of the head by using ImageJ software; positive cells in the eyes were removed from cell counts (TUNEL and phospho-histone H3). NIH ImageJ software was also used to measure the area of the proximal renal convolution or the cerebellum. Data were analysed and graphed using GraphPad Prism 7 software (GraphPad, San Diego, California, USA). Holm-Šídák multiple comparison testing following one-way analysis of variance was performed separately for each set of data corresponding to a phenotype. p Values less than 0.05, 0.01, 0.001 and 0.0001 are summarised with *, **, *** or ****, respectively. p Values >0.05 are indicated by ‘ns’ (non-significant).
Molecular cloning and site-directed mutagenesis
We obtained a Gateway-adapted full-length CEP55 open reading frame entry clone (Invitrogen clone IOH56860; NM_018131.4). We conducted site-directed mutagenesis to insert the c.171C>G (p.H57Q) or c.1133A>T (p.H378L) variants as described.20 The c.1274C>A (p.S425X) mutation was introduced by NorClone Biotech. The integrity of all clones was confirmed by restriction analysis and Sanger sequencing. The Gateway system (Life Technologies) was used to generate either monomeric Red Fluorescent Protein (mRFP)-tagged CEP55 constructs (wild-type (WT) or mutant) from pDest-733 for N-terminal fusion with mRFP (protein localisation studies); or template for SP6 in vitro transcription using pCS2+-dest (RNA injection into zebrafish embryos) according to standard protocols.21
Cell culture, transfections and subcellular localisation
COS-7 cells (CRL-1651 from ATCC), fibroblasts from a healthy individual (3348)22 and fibroblasts derived from infant 305 were grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% antibiotic-antimycotic (Gibco) in a humidified 37°C 5% CO2 incubator. We seeded COS-7 at 50–80% confluence on sterile glass cover slips in 6-well plates, incubated them for 24 hours and transfected with 1 μg of plasmid DNA for 48 hours using Effectene Transfection Kit (QIAGEN) according to the manufacturer's instructions. Cells were fixed in 3.7% paraformaldehyde in phosphate buffered saline (PBS; Gibco) for 20 min at room temperature, washed twice with PBS and incubated in 0.2% Triton X-100 (Sigma) in PBS for 5 min. Slides were blocked for 20 min using 2% bovine serum albumin (BSA) in PBS before both the primary and secondary antibody incubation. We incubated the slides with primary anti-α-tubulin antibody (rat monoclonal antibody, MCA77G, Cedarlane; 1:2000 in 2% BSA in PBS) for 1 hour at room temperature. We then used a goat α-Rat IgG (H+L) Alexa 488 antibody diluted 1:300 in 2% BSA in PBS incubated at room temperature for 30 min. The coverslips were washed three times with PBS, dipped in deionised water, inverted and mounted onto glass microscope slides with Vectashield with DAPI (Vector Laboratories) and sealed with nail polish. Microscopic imaging was performed on an Olympus BX51 fluorescent microscope (Olympus, Canada, ON, Canada) at 60× magnification with comparable exposure times for each experiment. Images were recorded with Northern Eclipse software (Empix, ON, Canada) and processed with Photoshop CS3 and Illustrator CS3 (Adobe).
Western blotting
48-hours post transfection, protein lysates were harvested from COS-7 cells. After a brief wash with PBS (pH 7.4), cells were harvested in hot (95°C) 1×SDS sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS and 10% Glycerol). We used QIAshredder columns (Qiagen) to homogenise samples according the manufacturer's instructions. Subsequently, we added reducing buffer (2×SDS sample buffer and 2-mercaptoethanol (Sigma) in a 1:1 ratio, supplemented with bromphenol blue dye (Sigma)). Samples were heated at 95°C for 5 min and placed on ice for 5 min. 40 µg of each protein lysate was analysed by SDS-PAGE (using a BioRad Mini-Protean TGX Stain-Free Gel) followed by western blotting using Immobilon-FL Polyvinylidene fluoride (PVDF) membranes (EMD Millipore). The membrane was first incubated with blocking buffer consisting of 5% milk powder in Tris buffered saline (TBS, Thermo Scientific) without Tween-20, and then incubated with anti-CEP55 (1:500; mouse polyclonal antibody raised against full-length human protein; H00055165-B01P; Abnova) in blocking buffer consisting of 5% milk powder in TBS with 0.1% Tween-20 at 4°C overnight. After washes with TBS with 0.1% Tween-20, blots were incubated with blocking buffer containing IRDye 800CW Goat anti-Mouse IgG (H+L) (1:15 000; Mandel Scientific) for 30 min at room temperature. Prior to imaging, blots were washed with TBS with 0.1% Tween-20, followed by a 5-min incubation in TBS. mRFP-CEP55 protein bands were detected with an Odyssey FC Imager (Li-Cor) and imaging was conducted with Image Studio V.5.0 software (Li-Cor).
Results
Clinical features
The family visited our genetics clinic with their first pregnancy after an ultrasound examination at 20 weeks gestation showed anhydramnios, small dysplastic kidneys and hydranencephaly. The mother was 25 years old and the antenatal history was unremarkable, with normal prenatal screening and no known teratogenic exposures. Both parents had Dutch-German Mennonite ancestry, suggesting a possible degree of relatedness, but they were not aware of any consanguinity. The family history was otherwise non-contributory. This fetus was stillborn at 30 weeks gestation (infant 303). Our fetal assessment unit followed three subsequent pregnancies from the first trimester. Two of these fetuses were also found to have abnormalities similar to the first fetus and were each delivered in the third trimester; one male died within minutes of birth at 35 weeks gestation (infant 305) and the other male was stillborn at 32 weeks gestation (infant 306).
Postmortem examination of all three affected children showed evidence of Potter sequence including microretrognathia, limb contractures, single transverse palmar creases, fifth finger clinodactyly, talipes equinovarus and pulmonary hypoplasia. We noted additional dysmorphisms that included a pinched nose with a bulbous tip, choanal narrowing, broad hands with a short fifth digit and cutaneous syndactyly of the toes with a wide first web space (see figure 1A and online supplement 2 for detailed pathologic description). The two infants who had full autopsies (303 and 306) showed dysplastic aortic valves, dilated left ventricles and only vestigial remnants of renal tissue with a few glomeruli in a background of collagenous tissue. The third infant (305) had an incomplete autopsy because advanced autolysis compromised tissue examination. All three had hydranencephaly with a normal head size relative to the rest of the body and cystic replacement of most of the neural tissue (figure 1B–D). The cerebellum, brain stem, basal nuclei and the basal portions of the temporal and frontal lobes were present, although hypoplastic. The bulk of the cerebral cortex was absent or reduced to islands of tissue attached to a leptomeningeal sac (see online supplementary figure S2.5A, B). To assess pathology further, we conducted microscopic examination; these analyses showed that the temporal cortex was disorganised. Approximately 5–10% of neurons and glial cells were multinucleated in all regions of the brain (figure 1E, F) as well as in retroperitoneal sympathetic ganglia adjacent to the kidney tissue (see online supplementary figure S2.10B) and the ganglion cell layer of the retina (see online supplemental figure S2.10E). Normal human brain, even at an early developmental stage, does not have such abnormal neurons, although they have been reported recently in association with a severe form of primary microcephaly caused by autosomal recessive mutations in CIT, a gene involved in cytokinesis.23 We did not appreciate any evidence of infection, inflammation or vascular obstruction.

Morphological and histological phenotypes of MARCH syndrome. (A) Postmortem photographs depicting consistent dysmorphology among the three infants. Features secondary to Potter deformation sequence are evident: microretrognathia, flattened face, fifth finger clinodactyly, single palmar creases, talipes equinovarus and proximal flexion contractures (not depicted here). Also evident is a pinched nose with a bulbous tip, anteverted nares, choanal narrowing, small palpebral fissures, epicanthal folds (infant 305), low set ears, shortened neck with redundant skin and microstomia. Infant 306 had a particularly small mouth with ankyloglossia. The hands show brachydactyly with tapered fingers and a shortened fifth finger. The feet show variable degrees of cutaneous (and in one case osseous) syndactyly with a wide first webspace. (B) In utero sonograms showing the progression of the hydranencephaly in infant 305. Gestational age is in the upper corner of each photograph. A cystic hygroma is highlighted by the white arrowhead. (C) Postmortem MRI scan of infant 305. Shown is a slightly off-sagittal view of the cranium depicting the almost complete absence of the cerebral hemispheres. The hypoplastic tissue in the frontotemporal region and from within the posterior fossa are evident. (D) Postmortem view inside the cranial vault of infant 305. The view is from an occipital opening and the lack of most neural structures is evident. The top of the cerebellum and brainstem is visible as is the remnants of the frontotemporal lobes. (E) NeuN immunolabelling of frontal lobe sections at 100× magnification. A multinucleated neuron is highlighted by the strong NeuN staining. Such neurons represent 5–10% of the neurons amongst all sections. (F) Glial fibrillary acidic protein (GFAP) immunolabelling of a frontal lobe section at 60× magnification. Multiple nuclei within a glial cell (astrocyte) are highlighted by black arrowheads. Multinucleated glial cells tended to be less common than multinucleated neurons in all sections.
supplement data
Several known syndromes have hydranencephaly as a rare feature, including Cousin syndrome (OMIM#260660), Neu-Laxova (OMIM#256520) or Roberts syndrome (OMIM#268300). However, hydranencephaly is the primary feature only in proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (OMIM#225790), microhydranencephaly (OMIM#605013) and hydranencephaly with abnormal genitalia (OMIM#300215). None of these coincide clinically with our patients' presentation due to the lack of vascular abnormalities in the brain; proportionate head size; normal genitalia; renal dysplasia and characteristic dysmorphic features. The closest match with our patients is a single case report of two male siblings with hydranencephaly and renal aplasia (OMIM#236500).24 Through further literature searches, we identified four more reports of infants with the combination of hydranencephaly and renal dysplasia/aplasia.24–28 These nine reported infants all had remarkably similar phenotypes to our patients (table 1); they all have hydranencephaly and eight of the nine have renal aplasia or severe renal dysplasia. One child had presumably functioning kidneys (given that he survived for more than 1 month), but it is not clear whether there was renal dysplasia since he was not investigated in this respect.26 He likely has the same disorder, given that his sister had the full phenotype. Of those patients with neuropathological examination, a high incidence of multinucleated neurons was described.24 ,25 At least five of the infants had unusual feet, including large first web spaces and/or syndactyly.24 ,26 Of the nine cases, two were singletons and seven were from families with two or three affected children each. Four of the nine reported cases were female. Combined with our family, these data argue strongly for an autosomal-recessive aetiology, particularly since our family is from a population with a strong founder effect, although we cannot definitively rule out alternative aetiologies such as an X-linked inheritance or multiple independent genetic effects.29 We propose that this phenotype be known as MARCH syndrome.
Clinical spectrum of MARCH syndrome
Gene identification
We obtained postmortem DNA samples from all three fetuses. An amnion sample from Infant 303 had a 46,XY karyotype at 400 band resolution. DNA from infant 305 that was extracted from cultured fibroblasts showed a normal chromosomal microarray, ruling out the possibility of significant copy number abnormalities (180K Oligoarray, CombiMATRIX Diagnostics, Irvine, California, USA). Given the high recurrence rate and degree of similarity between the affected children, we suspected a single-gene aetiology, probably through the inheritance of an ancestral Mennonite founder allele.
Given an expectation of either homozygosity by descent or X-linked inheritance, we used SNP genotyping to identify candidate regions. All four siblings had multiple regions of homozygosity. Total levels of homozygosity ranged from 2.5% to 4.0% of the genome, supporting our suspicion of distant relatedness due to founder effect. One region of 9.4 Mb on chromosome 10 showed segregation consistent with an autosomal-recessive disease model (figure 2A). In addition, there were two regions (28.7 and 3.2 Mb) on the X chromosome that showed segregation consistent with an X-linked disease model (figure 2B).

Genetic analysis of the MARCH syndrome pedigree. (A) Circular ideogram of the genome (Agile MultiIdeogram; http://dna.leeds.ac.uk/). The outer circle represents the ideograms of chromosomes 1–22. Each inner circle represents homozygosity data from an individual from the family as marked. Homozygous blocks are coloured in either grey (unaffected), red (affected but not present in all three affected siblings) or blue (present in all three affected siblings and not in unaffected). (B) Segregation of the X chromosome in the four male siblings. Areas that are common among the affected fetuses but not present in the unaffected are highlighted. (C) Exome sequencing data from infant 306. The filtering parameters used to sort through the variant data set are shown. The most likely candidate disease-causing mutation is highlighted in the grey rectangle. (D) Sanger sequencing data for the CEP55 variant, showing traces bearing the mutation segregating in the family under an autosomal-recessive paradigm.
Next we performed exome sequencing on one of the affected infants. Mean CCDS coverage was 141X with 92% of bases covered by >20 reads. We identified ∼147 000 variants that exceeded quality thresholds. Filtering for rare non-synonymous/splice variants (<0.1% based on variant frequencies from publicly available and in-house exome datasets) in the candidate regions identified (figure 2A, B), we narrowed the list to a single candidate (figure 2C): a homozygous nonsense mutation in CEP55 (rs201430235, c.1274C>A, p.S425X (NM_001127182.1)). This variant has been reported previously in a large-scale variant identification project in two separate individuals, both of whom were heterozygous (http://genome.sph.umich.edu/wiki/Exome_Chip_Design). The ExAC database reports an allele frequency of 0.0016% (January 2016). The variant maps to the final exon of the gene and is predicted to escape nonsense mediated decay, resulting in a truncation of 40 amino acids at the C-terminus.30 Orthologs from 31/32 primates and placental mammals show conservation of this region, especially three serine residues (S425, S428 and S436), which have all been shown to be phosphorylated, and are essential for the function of this protein.31 ,32 Sanger sequencing in the family confirmed that the variant showed segregation consistent with autosomal-recessive inheritance (figure 2D). In an effort to identify additional variants that could contribute to the phenotype, we analysed the exome data using relaxed variant filtering (not shown). We included all regions that segregate under a compound heterozygous model and the frequency cut-offs were increased to <1%. Under these criteria, we only identified a single additional candidate gene, R3HCC1L (homozygous rs147674187). Although we cannot exclude the possibility of involvement, it has not been implicated in human disease and the variant is unlikely to be pathogenic (CADD score 1.5).33 Thus, CEP55 remains the most likely candidate molecular driver of disease.
CEP55 expression
Most reported functional studies on CEP55 have been performed either in cell culture or in tumour tissue.31 ,32 ,34–38 To investigate the spatiotemporal expression of CEP55, with emphasis on the developing human brain, we interrogated the Brainspan transcriptome database for CEP55 mRNA levels in the human brain during different time points of gestation.39 The highest levels of CEP55 expression were in fetuses 8–12 weeks gestation, with a decline by 16 weeks and minimally detectable levels by 37 weeks, which continued into adulthood. This corresponds to the timing of asymmetric cell division to produce neurons from neural progenitor cells destined for the neocortex and basal nuclei.40 Structures with the highest expression included the ganglionic eminence and occipital cortex, followed by the dorsolateral prefrontal cortex. Lowest levels (approximately 1/3 of highest values) were in the cerebellum and inferolateral temporal cortex, which we note to be regions of the brain spared from the major destructive process in our patients.
We derived a fibroblast cell line from infant 305 and studied the expression of CEP55 (figure 3A). We conducted RT-PCR and detected CEP55 mRNA expression, although it was decreased compared with a normal control, suggesting reduced, but not extinguished, mRNA stability (figure 3B). There was no evidence of splicing abnormalities and the mutant transcript could be detected by sequencing (figure 3C).
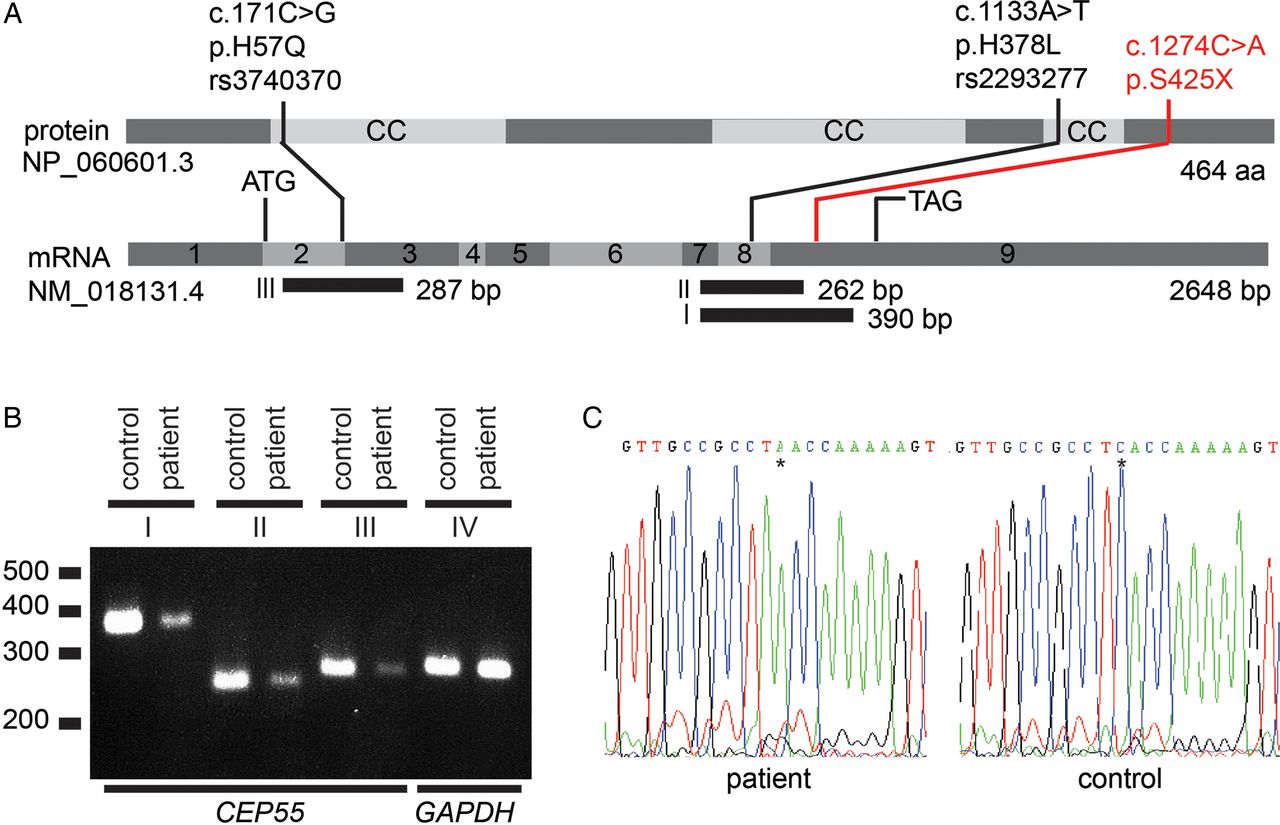
CEP55 expression studies in fibroblasts from a MARCH syndrome patient. (A) Schematic of CEP55. CEP55 mRNA transcript (NM_018131.4) and the encoded protein (NP_060601.3), indicating two polymorphisms (black) and the nonsense mutation detected in this study (red). The regions that were amplified using RT-PCR are indicated (fragments I, II and III). Polymorphisms rs3740370 and rs2293277 are common (allele frequencies of 0.23 and 0.70, respectively) based on the ExAC Browser (January 2016). (B) CEP55 mRNA expression is lower in patient-derived fibroblasts compared with control fibroblasts. Shown are RT-PCR fragments involving (I) exon 7–9 (390 bp), (II) exon 7–9 (262 bp), (III) exon 2–3 (287 bp) and (IV) control (GAPDH) expression (288 bp). cDNA input for each PCR was twofold in the patient compared with the control to equalise GAPDH expression. (C) Sanger sequences of fragment I of the patient and a healthy control from position c.1264 to c.1284. The asterisk indicates the position of the stop mutation c.1274C>A, resulting in p.S425X. There are no indications that this mutation induces aberrant splicing.
We attempted to perform immunohistochemical and western blot analysis of CEP55 in normal human tissue as well as samples obtained from the affected fetuses at autopsy using multiple commercial antibodies. We had significant difficulty obtaining reliable staining with most of these antibodies. We provide a detailed discussion of these experiments in online supplement 3 given the implications for future work involving CEP55.
supplement data
Modelling the effect of CEP55 truncation in vivo
To investigate the relevance of CEP55 disruption to the hallmark features of MARCH syndrome, we targeted the sole CEP55 ortholog in zebrafish (cep55l). A recent report showed that cep55l is expressed near-ubiquitously in developing zebrafish embryos, and that either transient suppression or stable mutation of cep55l leads to gross developmental defects likely due to altered Akt stability.6 Notably, these homozygous mutants display cell cycle dysregulation and massive apoptosis in anterior structures subsequent to the depletion of maternal transcript.6 To complement these studies, and to test the pathogenic potential of p.S425X, we first obtained two MOs that target the splice donor sites of cep55l exons 2 and 5 (sb1 and sb2, respectively; online supplementary figure S4.1A). First, we confirmed that both MOs reduce endogenous cep55l message. We injected 5 ng of each MO into zebrafish embryos at the one-to-two-cell stage, harvested RNA at 1 dpf and evaluated cep55l splicing near MO target sites with RT-PCR. For each of sb1 and sb2, we observed a reduction of detectable quantities of WT cep55l message in morphants, likely due to MO-induced aberrant splicing that introduced premature termination codons (see online supplementary figure S4.1B).
supplement data
Although cep55l mutant zebrafish display a constellation of gross anatomical abnormalities,6 we characterised further these defects in the cell and tissue types relevant to MARCH syndrome. We have shown previously that automated fluorescent imaging of live -1.4col1a1:egfp zebrafish larvae can demarcate structures in the pharyngeal skeleton, while enabling quantitative assessment of altered craniofacial morphology in zebrafish models of human disease.15 We injected zebrafish embryo batches with increasing doses of cep55l sb1 and sb2 and acquired ventral images of GFP-positive cells at 4 dpf (see online supplementary figure S4.1C). We observed a significant and dose-dependent increase in cartilage disorganisation, as quantified by the angle of the ceratohyal cartilage. Both MOs produced similar micrognathia phenotypes and could be rescued significantly with co-injection of WT human CEP55 message (figure 4A, B and online supplementary figure S4.1D, E; repeated, with masked scoring).

Transient suppression or CRISPR/Cas9 mediated genome-editing of cep55l results in craniofacial, neurological and renal abnormalities in zebrafish. (A) Representative live ventral views of 4 dpf -1.4col1a1:eGFP larvae showing craniofacial structures. cep55l F0 mutants or sb2 morphant larvae show significantly broader ceratohyal (CH) angles compared with controls. (B) Plot indicates the measurement of the ceratohyal angle (as shown in panel A). RNA injected alone shows no effect. Larvae injected with sb2 or sgRNA+Cas9 display significantly increased ceratohyal angle compared with controls. This phenotype can be rescued with coinjection of sb2 and wild-type (WT) (p.H378-RNA) or p.L378-RNA. sb2+S425X RNA does not rescue significantly. n=23–62 embryos/injection; repeated. (C) 4 dpf larvae stained for acetylated tubulin show hypoplasia of brain regions, including the cerebellum and optic tecta. Box and zoomed image indicate the cerebellum; asterisks (*) indicate optic tecta. (D) Plot indicates the quantification of the total area (µm2) of the cerebellum at 4 dpf, as indicated by orange outline in the control and sb2 insets of (C). n=31–81 embryos/injection; repeated. (E) Larvae fixed at 4 dpf and stained for Na+/K+ATPase are shown (lateral view; zoomed image represents the proximal convoluted tubule; outline indicates the area quantified in (F). Embryos injected with cep55 sb2 or sgRNA + Cas9 display proximal tubules of decreased area (µm2) and decreased presence of convolutions compared with control embryos. (F) Plot indicates the total area (µm2) of the proximal tubule in 4 dpf larvae; n=31–61 embryos/injection, repeated. *, **, *** and **** indicate p<0.05, 0.01, 0.001 and 0.0001 respectively. ns, not significant.
Next, we fixed and immunostained sb2 MO-injected larvae to evaluate brain structures at 4 dpf. Demarcation of the axon tracts using an anti-acetylated α-tubulin antibody16 ,41 ,42 revealed a significant reduction in the size of brain structures. Similar to the cerebellar hypoplasia observed in MARCH cases, we saw an ∼28% reduction in dorsal cerebellar area of cep55l morphants in comparison to controls (p<0.0001; figure 4C, D). To determine whether cep55l morphants display renal atrophy, we marked the renal tubules of 4 dpf larval batches with anti-Na+/K+ ATPase antibody. We observed a significant reduction in the lateral area and convolution of the proximal tubule in sb2 MO-injected batches (p<0.0001; figure 4E, F). Importantly, each of the brain and renal phenotypes could also be rescued significantly by co-injection of MO with capped human CEP55 mRNA. Finally, on the cellular level, we reproduced the previously reported and apparently normal M-phase marker staining (phospho-histone H3) with concomitant apoptosis in cep55l sb2 morphants (see online supplementary figure S4.2A–D and ref. 6).
To confirm further that the observed anatomical and cellular phenotypes were specific, we targeted the zebrafish cep55l locus with CRISPR/Cas9 genome-editing and repeated a subset of phenotyping measures. Consistent with the transient cep55l model, F0 mutants displayed altered craniofacial patterning with a significantly more obtuse ceratohyal angle than controls (p<0.0001), and atrophy of the proximal renal tubules (p=0.0016 vs. controls). A next-generation sequencing approach estimated that targeted mutation was present in as much as 76% of cells sequenced from 2 dpf embryos, but decreased to 17–25% by 4 dpf (see online supplementary figure S4.1F, G). The most likely explanation for the reduction is apoptosis in cells with cep55l disruption, which is supported by TUNEL assay at 2 dpf (see ref. 6 and online supplementary figure S4.2C, D).
Prompted by the observations that CEP55 mRNA harbouring p.S425X was detectable by RT-PCR from patient tissue and is predicted to produce a truncated protein, we carried out in vivo complementation assays to determine variant pathogenicity. First, we confirmed our assay specificity by comparing the rescue efficiency of human WT message (encoding p.H378) to that of message encoding a common variant at the same amino acid position, which was also identified in the affected fetuses (p.L378; rs2293277, allele frequency 0.70 in ExAC). For each of the craniofacial (ceratohyal angle measurement) and cerebellar area phenotyping approaches, we observed that CEP55 p.L378 also rescued the sb2 MO defects significantly (p<0.0001; both phenotypes, respectively; figure 5B, D). However, co-injection of sb2 MO with CEP55 mRNA harbouring the p.S425X variant produced jaw phenotypes that were significantly worse than WT mRNA (p.H378 or p.L378) but not significantly different from MO alone (figure 4B). The trend was similar in the cerebellar assay, but significant only for the p.L378 encoding mRNA (figure 4D). Taken together, our cep55l transient and F0 mutant data show relevance of gene disruption to the primary clinical features of MARCH syndrome and indicate that the CEP55 is a functional null variant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
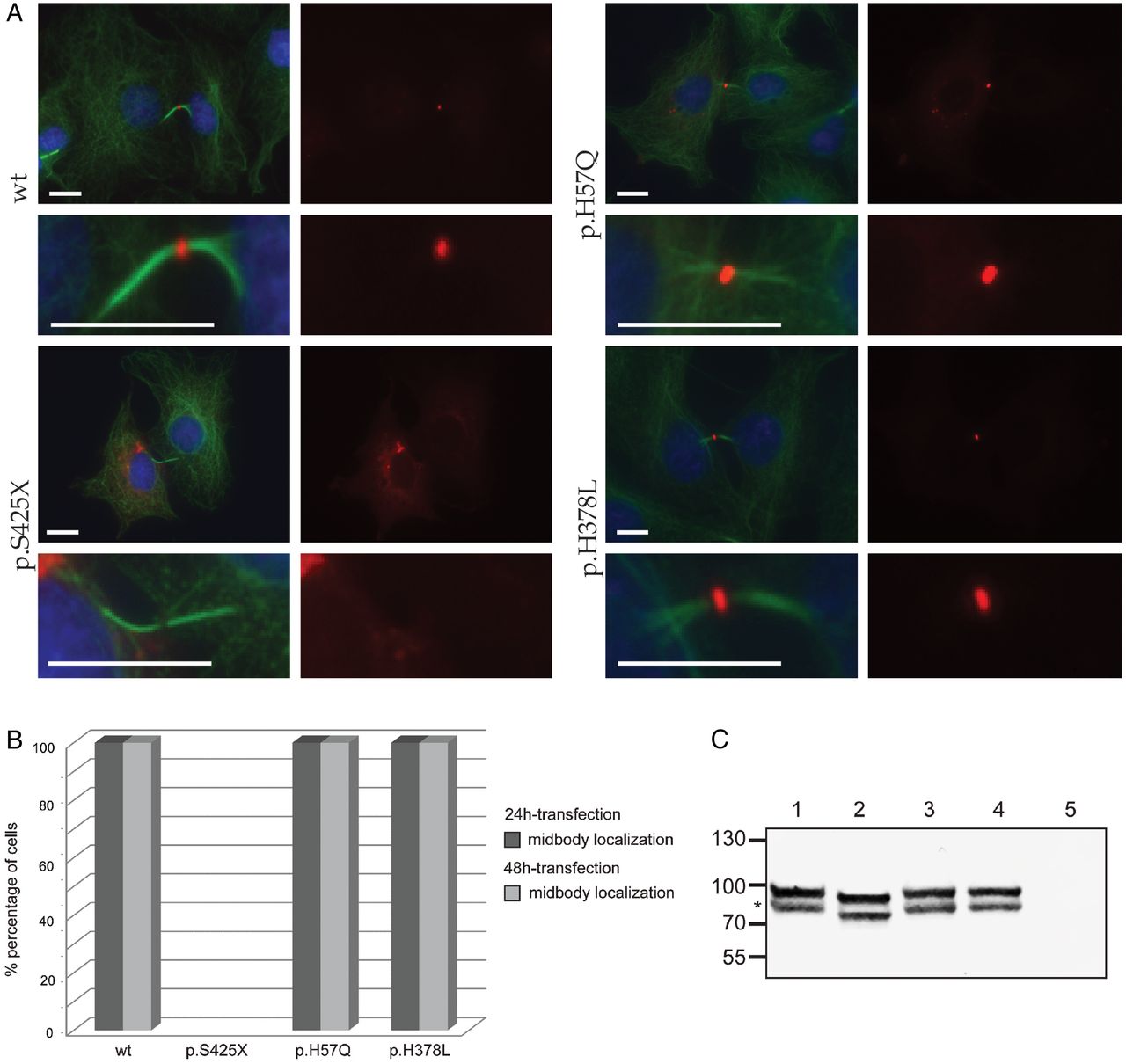
Subcellular localisation of CEP55. (A) Confocal images show midbody localisation of wild-type and polymorphism (p.H57Q or p.H378L) containing monomeric Red Fluorescent Protein (mRFP)-CEP55 in COS-7 cells. Mutant mRFP-CEP55 (p.S425X) did not localise to the midbody in transfected cells. A representative image of dividing cells is shown for each condition, and a magnification of the intracellular bridges is provided underneath each image. α-tubulin (green) marks mitotic bridges and nuclei were stained with DAPI (blue). mRFP-CEP55 is shown in red. Scale bars represent 10 μm. (B) Histogram showing the localisation of mRFP-tagged CEP55 proteins in COS-7 cells following 24 and 48-hour transfections, respectively. Wild-type mRFP-CEP55 and mRFP-CEP55 with polymorphisms p.H57Q or p.H378L localised to the midbody near the end of cytokinesis, while the mutant (p.S425X) did not. The differences between wild-type, polymorphisms and mutant conditions were significant (p<0.0001). The number of mitotic cells that were counted for the 48-hour transfection are n=52 (wild-type), n=51 (p.S425X), n=50 (p.H57Q) and n=50 (p.H378L). The number of mitotic cells analysed in the 24-hour transfection are n=79 (wild-type), n=52 (p.S425X), n=62 (p.H57Q) and n=69 (p.H378L). The 24-hour and 48-hour transfections were performed in duplicate. (C) Western blot analysis of mRFP-CEP55 in COS-7 cells. Lower bands (indicated by *) show the expected sizes: 81 kDa for mRFP-fused wild-type CEP55 and CEP55 with polymorphisms, and 77 kDa for the mutant protein. No CEP55 expression was detected in cells transfected with the empty vector. The origin of the higher band evident in the first four lanes is unclear. One potential explanation is a post-translational modification of the tagged CEP55 occurring in COS-7 cells.
Subcellular localisation
Previous studies have shown that CEP55 localises to the centriole during interphase and to the midbody on the intracellular bridge between two daughter cells during cytokinesis.43 Abnormal phosphorylation at the C-terminus of the protein prevents the association with the midbody and causes abscission failure.31 The mutation identified in our patients (c.1274C>A; p.S425X) is predicted to truncate the protein, causing a loss of all three key phosphorylated serines. We hypothesised that the patient mutation would lead to a similar mislocalisation of CEP55. To test this idea, we transfected COS-7 cells with four different full-length cDNA CEP55 constructs that contain an mRFP tag. These constructs encode WT mRFP-CEP55, mutant mRFP-CEP55 (p.S425X) or mRFP-CEP55 with a common polymorphism (p.H57Q, minor allele frequency 0.23 in ExAC; or p.H378L) (figure 5A, B). At 24 hours or 48 hours post transfection, we examined protein localisation. As expected, WT and polymorphism-containing mRFP-CEP55, but not the S425X-encoding mutant localise to the midbody in dividing cells. On western blot with anti-CEP55 antibody, either WT or polymorphism-containing mRFP-CEP55 migrate to the expected size (81 kDa), while mutant CEP55 is truncated (77 kDa; figure 5C); these observations support our imaging studies and together suggest that the last 40 amino acids of CEP55 are essential for targeting CEP55 to the midbody of dividing cells.
Discussion
Here we report the identification of a homozygous truncating mutation in CEP55 as the likely cause of a new perinatal lethal syndrome we term MARCH. The genetic data that led to the identification of the CEP55 mutation is from a single family, and therefore, it is difficult to rule out the possibility of multiple genetic effects leading to this phenotype. However, our functional data strongly support CEP55 as the sole contributor to this complex condition.
CEP55 encodes, a 464 amino acid peptide that contains three central coiled-coil domains.31 CEP55 is an essential regulator of the final stages of mitosis; in cultured fibroblasts, the protein localises to the centrosome and pericentriolar matrix during most of the cell cycle but diffuses throughout the cell at the onset of mitosis (prophase). Later in mitosis (anaphase/telophase) CEP55 aggregates at the midzone, where cell separation will eventually take place. During cytokinesis, it is concentrated in the midbody, the protein complex present in the cytoplasmic bridge between the two daughter cells.43 There, it recruits the Endosome Sorting Complex Required for Transport machinery, and other components involved in membrane fission, to promote the formation of the cleavage furrow and eventually induce abscission of the two cells.44 The disassociation of CEP55 from the centriole appears to be dependent on phosphorylation of the two C-terminal serines, p.S425 and p.S428, by the protein kinases Erk2 and Cdk1.31 Subsequently, myotubularin proteins connect CEP55 to another protein kinase, PLK1, which phosphorylates CEP55 at p.S436. This stabilises the protein and leads to its orderly introduction into the midbody.32 ,38 ,45 ,46 If either of these phosphorylation events does not occur then cytokinesis fails with mitosis taking place but the daughter cells being unable to separate, leading to a binucleated cell.31 ,32 ,34
We show that the nonsense mutation (c.1274C>A; p.S425X) identified in our family likely truncates CEP55. In cell culture experiments, the truncated version of CEP55 does not localise to the midbody during cytokinesis. Given the loss of the three phosphorylated residues, our data suggest the presence of multinucleated cells in the affected fetuses is a consequence of failed cytokinesis. We also show that cep55l knockdown and CRISPR/Cas9 mutation in zebrafish recapitulates most key features of the syndrome: we reproduce the cellular apoptosis and neurological abnormalities reported previously in zebrafish F2 mutants harbouring a homozygous c.115C>T; p.E39X mutation;6 we quantify further the cerebellar, renal and craniofacial features of our cep55l zebrafish models; and show that these phenotypes can be rescued by full length, but not truncated human CEP55 mRNA. It is possible that the lack of overt hydranencephaly in zebrafish morphants or mutants is due to interspecies differences, particularly with regard to brain development and anatomy.
The mechanism by which this nonsense mutation in CEP55 ultimately leads to such a dramatic human neurological phenotype remains incompletely understood. We know that there are large numbers of multinucleated cells in what remains of the patient brain tissue, suggesting that abscission failure likely plays some part in the creation of this anomaly. It is not clear how the production of multinucleated neurons along with a potential migration abnormality would induce such widespread cell death in the cerebral hemispheres. One potential mechanism is through suppressed PI3K/AKT signalling as this pathway has been found previously to be downregulated in zebrafish with absent CEP55 function.6 Previous work has shown that activation of the PI3K/AKT pathway protects against cerebral ischaemic damage in animal models and protects cultured neurons against hypoxic and excitotoxic injury.47–49 A similar pathway is involved in ischaemic damage to kidney.50 Deficient PI3K/AKT signalling may thus help explain the destructive features in the brains of the fetuses affected with MARCH. Interference with the PI3K/AKT signalling pathway by virtue of mutant CEP55 could allow ischaemic-type damage in vulnerable organs, such as the parts of the brain with distal circulation, thereby leading to destruction of the cerebrum with resultant hydranencephaly.
In summary, we report a novel human phenotype that combines hydranencephaly, renal dysplasia, multinucleated neurons, syndactyly and characteristic dysmorphism that we have named MARCH syndrome. We provide evidence that it is caused by autosomal-recessive loss-of-function mutation in CEP55, a gene not associated previously with human disease. Our data provide insight into the developmental functions of CEP55 particularly as it relates to brain development. Future directions in our research include exploring the role of CEP55 in the creation of new neurons and understanding how PI3K/AKT may be involved in hydranencephaly in this syndrome and other conditions that include this anomaly. We propose that both measuring AKT activity in animal models of this disorder, and also testing the hypothesis that amelioration of AKT signalling might improve some of the observed neuroanatomical defects will provide further insight into the basis of this syndrome.
supplement data
Acknowledgments
The authors thank the family for agreeing to allow them to publish this research. They also acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Genome Quebec Innovation Centre and the Centre for Applied Genomics, Toronto, Canada, for SNP genotyping. They further thank H. Almadhoun, S.M. Farhan, J. Wang, M. Ban, J. Robinson, J. Samsoondar, C. O'Neil, V. Ly, and X. Mao for technical assistance and/or discussions, S. Phillips for postmortem examination, R. Roepman for vectors, G. Pickering for use of laboratory equipment, V.M. Siu for her ethics contribution, and L. Gyenis and D.W. Litchfield for scientific input.
References
Footnotes
Collaborators FORGE Canada Consortium (FORGE Steering Committee membership is listed in Supplement 1) Canadian Rare Diseases: Models & Mechanisms Network (Canadian Rare Diseases: Models & Mechanisms Network Committee memberships are listed in Supplement 1).
Contributors PF and HHA contributed equally. PF provided clinical care, designed the study, performed homozygosity mapping and wrote the initial manuscript. HHA and RH performed confocal microscopy, RT-PCR, aided in study design and contributed to manuscript preparation. JP, CSG, CR and YPL performed zebrafish experiments and contributed to manuscript preparation. ELB performed confocal microscopy, RT-PCR, and contributed to manuscript preparation. BC provided clinical care and contributed to manuscript preparation. LS performed variant validation and contributed to manuscript preparation. JM and SF provided bioinformatics support and contributed to the manuscript preparation. FORGE and RDMM are consortia that supported the study and members were involved in gene identification, connecting the collaborating authors and providing feedback on the manuscript. NK and EED designed the zebrafish experiments, aided in overall study design and contributed to manuscript preparation. CG and MRDB performed neuropathological analysis, immunohistochemistry, western blotting and contributed to manuscript preparation.
Funding This work was supported in part by the FORGE Canada consortium and a catalyst grant from the Canadian Rare Diseases: Models & Mechanisms Network (RDMM). Both of these initiatives are funded through grants from the Canadian Institutes of Health Research (CIHR) and Genome Canada with additional funding for FORGE being provided by the Ontario Genomics Institute, Genome Quebec, Genome British Columbia, and the McLaughlin Centre. This work was also supported by US NIH grants DK096415, DK075972 and HD042601 (NK); DK072301 and DK096493 (NK and EED). PF received support from the Rare Disease Foundation and the B.C. Children’s Hospital Foundation (P#46). HHA also received support from the Netherlands Organization for Scientific Research (NWO Veni-91613008) and the Dutch Kidney Foundation (CP11.18 ‘KOUNCIL’). MRDB is supported through the Canada Research Chair in Developmental Neuropathology. CSG was supported by a summer studentship from the Children’s Hospital Research Institute of Manitoba. NK is a distinguished Jean and George Brumley Professor.
Competing interests None declared.
Patient consent Obtained.
Ethics approval University of Manitoba. University of Manitoba, University of Ottawa, University of Western Ontario, and McGill University.
Provenance and peer review Not commissioned; externally peer reviewed.