Article Text

Abstract
Oral–facial–digital syndromes (OFDS) gather rare genetic disorders characterised by facial, oral and digital abnormalities associated with a wide range of additional features (polycystic kidney disease, cerebral malformations and several others) to delineate a growing list of OFDS subtypes. The most frequent, OFD type I, is caused by a heterozygous mutation in the OFD1 gene encoding a centrosomal protein. The wide clinical heterogeneity of OFDS suggests the involvement of other ciliary genes. For 15 years, we have aimed to identify the molecular bases of OFDS. This effort has been greatly helped by the recent development of whole-exome sequencing (WES). Here, we present all our published and unpublished results for WES in 24 cases with OFDS. We identified causal variants in five new genes (C2CD3, TMEM107, INTU, KIAA0753 and IFT57) and related the clinical spectrum of four genes in other ciliopathies (C5orf42, TMEM138, TMEM231 and WDPCP) to OFDS. Mutations were also detected in two genes previously implicated in OFDS. Functional studies revealed the involvement of centriole elongation, transition zone and intraflagellar transport defects in OFDS, thus characterising three ciliary protein modules: the complex KIAA0753-FOPNL-OFD1, a regulator of centriole elongation; the Meckel-Gruber syndrome module, a major component of the transition zone; and the CPLANE complex necessary for IFT-A assembly. OFDS now appear to be a distinct subgroup of ciliopathies with wide heterogeneity, which makes the initial classification obsolete. A clinical classification restricted to the three frequent/well-delineated subtypes could be proposed, and for patients who do not fit one of these three main subtypes, a further classification could be based on the genotype.
- oral-facial-digital syndromes
- ciliopathies
Statistics from Altmetric.com
Introduction
Oral–facial–digital (OFD) syndromes are rare genetic disorders characterised by the association of abnormalities of the face (hypertelorism and low-set ears), oral cavity (lingual hamartoma, abnormal frenulae and lobulated tongue) and extremities (brachydactyly and polydactyly). OFD syndromes also comprise a broad range of additional features that initially led to the clinical delineation of 13 OFD subtypes with mainly OFDI (polycystic kidney disease and corpus callosum agenesis), OFDIV (tibial dysplasia), OFDVI (mesoaxial polydactyly, vermis hypoplasia and molar tooth sign (MTS)) and OFDIX (retinopathy).1 2 More recently, a new subtype has been described associated with microcephaly which has designated OFDXIV by OMIM (MIM 615948). However, the precise phenotypic description revealed new unclassified OFD subtypes, in particular with severe microcephaly.3–6 Classically, the inheritance pattern is autosomal recessive except for OFDI, which has dominant X-linked inheritance and is lethal in males. Until recently, the molecular bases of OFD syndromes were poorly known. A few years ago, the OFD1 gene [MIM 300170] was initially described as causing the OFDI subtype.7 OFD1 encodes a protein located in the centrosome and basal body of primary cilia, suggesting that OFD syndromes are ciliopathies.
Ciliopathies are human diseases defined by ciliary structural and/or functional defects. Cilia, microtubule-based organelles projecting from the cytoplasmic membrane of the cell body, are divided into motile and non-motile or primary cilia. The primary cilia appear to be essential in several biological processes especially during development8 and serve a broad range of specific sensory processes using receptors and ion channels to sense photo, chemo and mechanical stimuli and allow the transduction of signalling pathways. Four structural compartments have been described1: the centrosome, composed of two centrioles (mother and daughter) and pericentriolar material, including the mature mother centriole, which converts to the basal body that orients and positions the cilium2 9; the basal body formed where the centrosome, a microtubule-organising centre, migrates to the cell surface to initiate cilium assembly3; the transition zone (TZ), located at the distal end of basal body and composed of Y-links connecting microtubules to the ciliary membrane and ciliary necklace and4 the transition fibres, which form the ciliary gate and constitute a diffusion barrier to regulate cytoplasmic protein entry into the ciliary compartment.10 11 The microtubules extend distally from the basal body to form the axoneme, where receptors localise on the apex and the ciliary membrane, a lipid bilayer distinct from the plasma membrane, and surround the cilium. Proteins are transported along the axoneme to permit ciliary growth, maintenance and function. This essential intraflagellar transport is composed of two modules: IFT-A for retrograde transport and IFT-B for anterograde transport, which distribute ciliary molecules to the different ciliary compartments.12
Ciliopathies present a broad range of features (retinopathy, cerebral malformations, bone defects, deafness or renal disease, and others); they are thus highly genetically heterogeneous diseases and include nephronophthisis (NPHP), Joubert syndrome (JBS), Meckel-Gruber syndrome (MKS), Bardet-Biedl syndrome (BBS) and different chondrodysplasias. Multiple allelism has been described, suggesting that human ciliopathies are genetically complex.13 More recently, mutations in six additional genes that encode ciliary proteins have been identified in one or two patients with OFDS: centrosomal proteins implicated in centriole elongation (NEK1 [MIM 604588], SCLT1 [MIM 611399] and TBC1D32/C6orf170 [MIM 615867]), proteins located in the TZ (TMEM216 [MIM 613277] and TCTN3 [MIM 613847]) and a protein that regulates ciliary signalling (DDX59 [MIM 615464]). Each known gene appears to be implicated in a classified OFD subtype: OFD1 in OFDI [MIM 311200] with polycystic kidney disease and corpus callosum agenesis, TCTN3 in OFDIV [MIM 258860] with tibial defect, DDX59 in OFDV [MIM 174300], TMEM216 in OFDVI [MIM 277170] characterised by cerebellar hypoplasia with the MTS and SCTL1 and TBC1D32/C6orf170 in OFDIX [MIM 258865] with coloboma.7 14–22
Using a strategy of whole-exome sequencing (WES), we identified five new causal genes in OFD syndromes and showed the implication of four additional genes previously reported in other ciliopathies as well as their different ciliary functions. In this unique cohort, all novel genes have been published independently. This paper presents an overview of the whole series and discusses the classification of this group with the advance of molecular delineation.
Patients and methods
Patient cohort
We gathered an international cohort of 115 index cases affected with different OFD syndromes. In all cases with a typical OFD I phenotype, we looked for OFD1 SNV or CNV by Sanger sequencing and targeted array-CGH, respectively.23 24 Causal OFD1 SNV or CNV were identified in 59/115 cases. Among the 56 other index cases with atypical clinical features or negative OFD1 sequencing (figure 1 and online supplementary table S1), we performed WES in 24 index cases, including 17 sporadic cases and 7 cases from consanguineous parents. WES was limited to 24 cases because of the quality and quantity of patients’ DNA and the availability of parental DNA. All of the patients presented oral abnormalities (lingual hamartoma, abnormal frenulae or lobulated tongue), facial dysmorphism and extremity abnormalities (mainly polydactyly), associated with cerebral malformations (12/14 cases), retinopathy (3/16 cases), renal abnormalities (4/14 cases) and/or cardiac malformations (9/17 cases). Six individuals were diagnosed with OFDVI because of the MTS on brain MRI and positive diagnostic criteria, two with OFDII and one with OFDV.25 Parental DNA samples were available in 17/24 cases.
Supplementary file

Clinical pictures, X-rays and brain MRI of cases with oral–facial–digital syndrome. Case 3a (K), case 3b (L), case 4 (B, N, V), case 5 (κ), case 6a (A, R, S, T, U), case 6b (F, Y, Z, α, β), case 7 (G), case 8 (E, L, Q, ε, ζ, ι, κ), case 10 (J, Y), case 11 (O), case 17 (D, I, K), case 19 (E), case 22 (ε), case 25 (λ, μ), case 26b (D, X, ζ), case 27 (P, ν, ο, π) case 28b (Q, ρ, ς), case 29 (υ,φ) with facial dysmorphism (A–D) including low-set ears, median pseudo-cleft of upper lip (F), missing incisors (A) or severe microcephaly (B), abnormal frenulae (E), cleft palate (I), lobulated tongue or hamartoma (G, H, J), pre and postaxial polydactyly of hands and feet (R, S, V, W, ε, ζ, ι, κ-υ), broad duplicated and/or deviated hallux (T, U, V, ε, ζ, η, θ, μ, ν, υ), Y-shaped metacarpal abnormality (κ, π), hypothalamic hamartoma (P), cerebellar hypoplasia (Q), brain MRI with MTS (K–O).
Exome analysis
After written consent had been obtained, blood samples were collected and DNA was extracted. Three micrograms of genomic DNA per index individual was subjected to whole-exome capture and sequencing using the SureSelect Human All Exon V5 kit (Agilent). The resulting libraries were sequenced on a HiSeq 2000 (Illumina) as paired-end 76 bp reads. Binary Alignment Map files were aligned to a human genome reference sequence (GRCh37/hg19) using BWA (Burrows-Wheeler Aligner; v0.6.2). All aligned read data were subject to the following steps: (1) duplicate paired-end reads were removed by Picard 1.77, (2) indel realignment and (3) base quality score recalibration were done on the Genome Analysis Toolkit (v2.1–10). Variants with a quality score >30 and an alignment quality score >20 were annotated with SeattleSeq SNP Annotation (see Web resources). CNV were detected by XHMM software (https://www.atgu.mgh.harvard.edu/) and annotated using chromosomic coordinates of coding exonic sequences on the human genome (https://www.ncbi.nlm.nih.gov/refseq/). Rare variants present at a frequency above 1% in dbSNP 138, ExAC Browser and the NHLBI GO Exome Sequencing Project or present in 312 exomes of unaffected individuals were excluded (see Web resources). To improve our exome analysis, data were crossed with a list of known cilia-related genes from the Ciliome Database, Cildb v2.1, Syscilia (see Web resources) and transcriptomic, proteomic and bioinformatics studies of cilia to identify putative ciliary genes.26–29 First, we looked for SNV or CNV in the six known genes in OFDS (OFD1, TCTN3, TMEM216, SCLT1, TBC1D32/C6orf170 and DDX59). We then focused on genes with homozygous variants in consanguineous families and with two heterozygous variants in other cases and prioritised (1) genes associated with human disease in ClinVar or HGMD databases (see Web resources), (2) cilia-related genes and (3) other genes (figure 2).
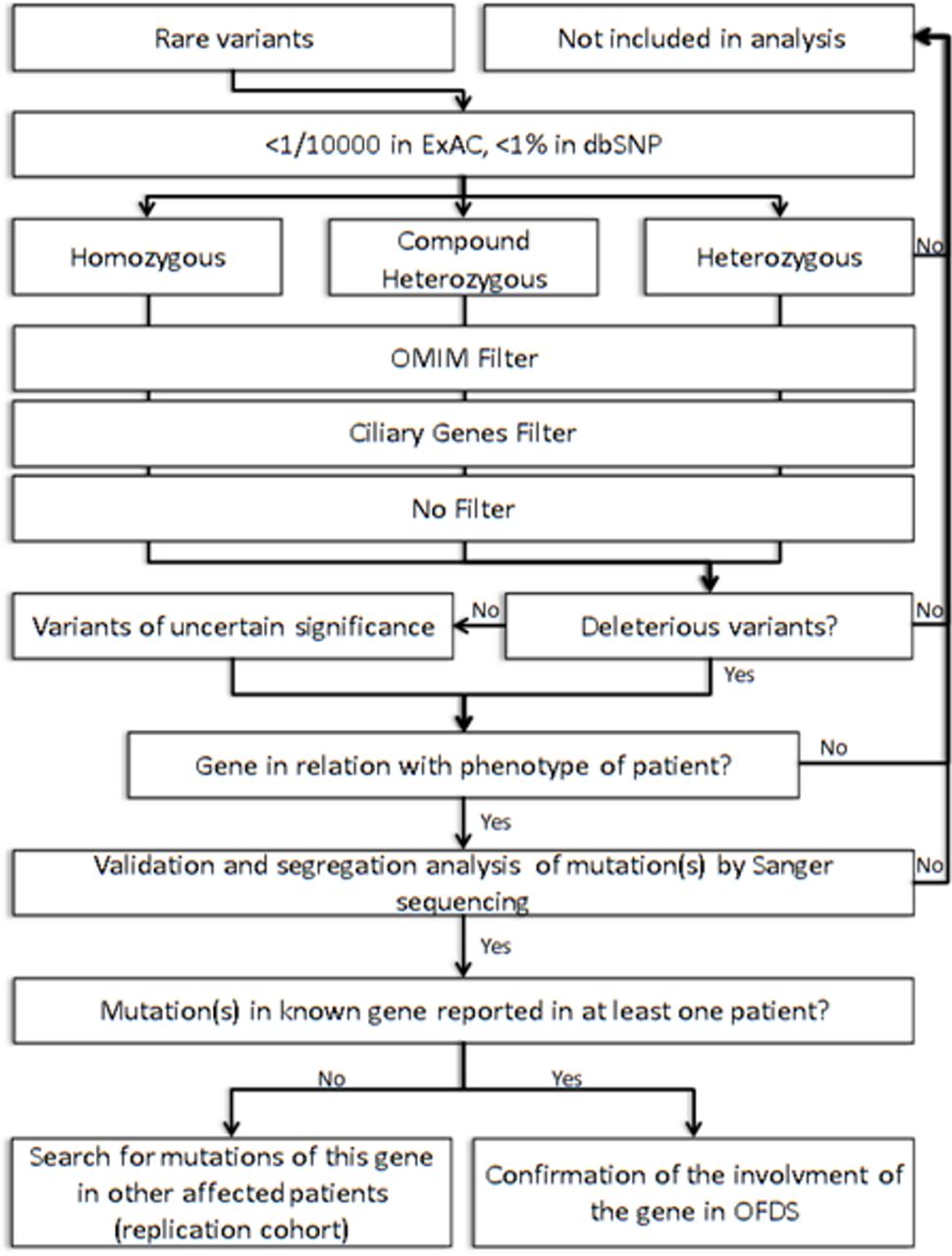
Strategy for exome analysis. dbSNP, SNP database; OFDS, oral–facial–digital syndrome.
Sanger sequencing
Candidate variants and parental segregation were confirmed by Sanger sequencing. The different primers are available on request. Genomic DNA was amplified by PCR using HotStarTaq PCR kit (Qiagen) according to the manufacturer’s protocol. PCR products were purified by the Agencourt CleanSEQ system (Beckman Coulter) and sequenced with the BigDye Terminator Cycle Sequencing kit, v3.1 (Applied Biosystems) in ABI 3730 sequencer (Applied Biosystems). Sequence data were analysed using Mutation Surveyor v4.0.9 (Softgenomics).
Results
WES identified causal mutations in 14/24 cases. The first analysis of known genes implicated in OFDS identified a homozygous missense variant in the DDX59 gene (MIM 610621) and heterozygous mutations in the OFD1 gene (MIM 311200) in three unrelated cases (p.Tyr87Cys, p.Ala614Hisfs*15 and c.655-8A>G, predicted to affect a splice site). In these latter cases, OFD1 mutations were not previously detected by Sanger sequencing.
The filtering strategy extracted five homozygous variants in consanguineous families (table 1): a frameshift in the INTU gene (MIM 610621), a non-sense mutation in the C2CD3 gene (MIM 615944), TMEM138 (MIM 614459) and TMEM107 genes, and a synonymous variant affecting a splice site in the IFT57 gene (MIM 606621).6 30–33 For all these genes, Sanger sequencing and parental segregation confirmed the homozygous status in the affected cases and the heterozygous status in each parent. We also identified compound heterozygous variants in four ciliary genes (table 1): TMEM231 (MIM 614949), WDPCP (MIM 613580), C5orf42 genes (MIM 614571) and KIAA0753.31 33–35 Sanger sequencing and parental segregation confirmed the compound heterozygous status in the affected cases and the heterozygous status in each parent for all genes, except for the KIAA0753 gene. For this gene, Sanger sequencing confirmed that the non-sense variant (NM_014804.2:p.Lys631*) was maternally inherited and the intronic substitution (NM_014804.2:c.1546–3C>A) occurred de novo and affected a splice site causing a truncated protein.34
Oral–facial–digital (OFD) genes identified by whole-exome sequencing or single-gene sequencing
The clinical heterogeneity of OFD syndromes was confirmed with various atypical signs and the overlap between OFD subtypes. Patients with a mutated OFD1 gene presented typical signs of the OFDI subtype (lingual hamartoma, lobulated or bifid tongue, cleft palate, renal disease and corpus callosum agenesis) associated with very rare abnormalities including cardiac malformations (case no 20), the MTS on brain MRI (case no 13) or 11 pairs of ribs (case no 12), which suggest overlapping with other subtypes. Variants in TMEM138, TMEM107 and C5orf42 caused OFDVI, characterised by the MTS. In unclassified OFD, C2CD3 mutations were associated with severe microcephaly, INTU and WDPCP mutations with cardiac defects, and IFT57 mutations with chondysplasia. DDX59 mutations had previously been reported in OFDII and identified in this cohort in a case of OFD V (no 1). OFDV, characterised by a median cleft of the upper lip and post-axial polydactyly, overlapped with OFD II, but this was predominantly found in patients of Indian origin. Finally, variants in the TMEM231 gene were identified in a foetal case with unclassified OFD.
We thus identified causal mutations in five new genes, in four genes previously implicated in other ciliopathies and in two genes previously known to be responsible for OFD syndromes (figure 3).

Localisation of proteins encoded by the 16 oral–facial–digital (OFD) genes in primary cilia. Five new OFD genes (in red), four genes previously implicated in other ciliopathies (in green) and seven genes previously reported in OFD: two with presented mutations (blue) and five others (white).
Discussion
This study presents the largest OFD cohort investigated by WES. It led to the identification of causal mutations in 58% of affected cases, thus confirming the power of WES in identifying the genetic cause in well-phenotyped cases and highly heterogeneous disorders.
Wide clinical and genetic heterogeneity of OFD syndromes
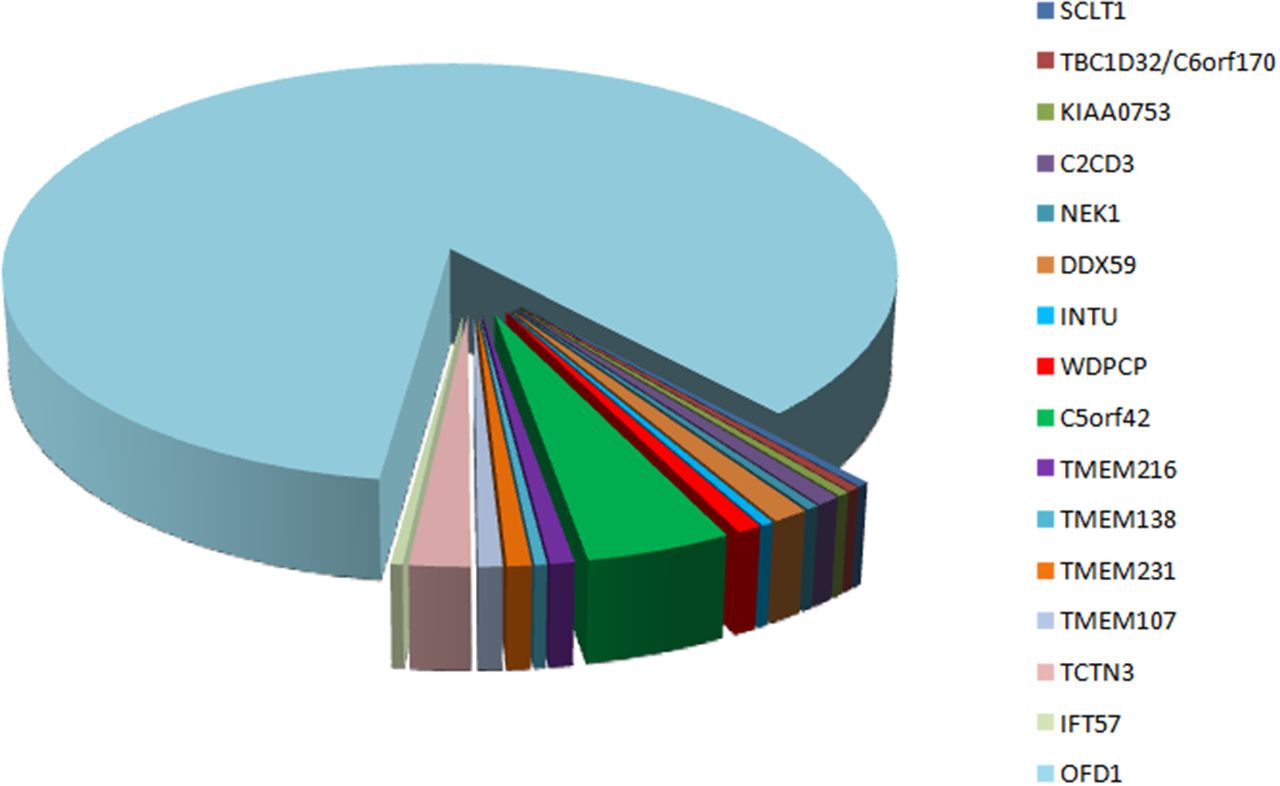
The wide clinical heterogeneity and variable modes of inheritance in OFD syndromes suggest extreme genetic heterogeneity. Exome sequencing thus appeared the obvious choice, and because OFD syndromes were suspected to be mainly recessive, we initially focused on homozygous or potential compound heterozygous mutations and prioritised ciliary genes and truncating rare variants in the absence of OMIM genes. In cases of suspected consanguinity, the probable causal variant was expected to be located within a large stretch of a homozygous region, thereby making it easier to identify new genes. Causal variants were thus identified in five new genes, at the homozygous status (C2CD3, INTU, IFT57 and TMEM107) or compound heterozygous status (KIAA0753).6 32 33 Recently, additional C2CD3, TMEM107 and TMEM231 mutations confirmed the implication of these genes in OFD syndromes (table 1).36 37 Causal variants were also identified in six other genes previously implicated in OFD syndromes (DDX59 and OFD1) or in other ciliopathies (TMEM138, C5orf42, TMEM231 and WDPCP). In all these patients, the OFD phenotype was clinically heterogeneous with OFDI (OFD1), OFDV (DDX59), OFDVI (TMEM138, TMEM107, KIAA0753, OFD1 and C5orf42) or OFDXIV (C2CD3) as well as unclassified OFD (TMEM231, IFT57, INTU and WDPCP), with cerebellar hypoplasia, severe microcephaly, chondrodysplasia or cardiopathy. These results demonstrate the wide clinical and genetic heterogeneity of OFD syndromes with, to date, 16 different genes. However, except for OFD1, few mutations have been reported in the other OFD genes, because OFD syndromes remain rare with wide genetic heterogeneity and because some mutations are found in specific ethnic groups (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Distribution of mutated genes in genotyped oral–facial–digital cases reported in this study and in the literature.
Frequent clinical and genetic allelism between OFD and ciliopathies
The progressive identification of the molecular bases has highlighted the involvement of the primary cilia in OFD syndromes and confirmed the clinical and genetic overlap between OFD and other ciliopathies.38 Indeed, OFD1, which is responsible for OFDI syndrome, was also reported in JBS and severe retinitis pigmentosa.39–42 TMEM216, initially implicated in JBS and MKS, also caused OFDVI.43 Moreover, we identified OFD mutations in the TMEM107 gene, which also causes JBS30 33 as well as in four other genes previously implicated in other ciliopathies (TMEM138, C5orf42, TMEM231 and WDPCP) (table 1). To date, allelism with other ciliopathies affects nine of the 16 OFD genes. The most frequent allelism concerns OFDVI and JBS (TMEM216, TMEM138, TMEM231, TMEM107, OFD1 and C5orf42).30 31 33 35 43 44 TMEM231, TMEM107 and C5orf42 genes also cause MKS,30 45 46 thus confirming the clear allelism between OFDVI, MKS and JBS syndromes with variable phenotypic expression. INTU and WDPCP mutations are also reported in NPHP and BBS, respectively, but the allelism between OFD and BBS remains uncertain because of the absence of clinical data in the reported cases.33 47 Recently, C2CD3 mutations have also been reported in two foetuses with skeletal dysplasia, suggesting a short rib-polydactyly (SRP) syndrome.48
Characterisation of three ciliopathy protein complexes and cilia disturbance in OFD syndromes
The clinical description of different subtypes suggested that the causal proteins could be assembled in different functional modules. Because the 16 genes encode for proteins located in different compartments of primary cilia, new ciliary functions were suspected of being implicated in OFD syndromes (table 2). Different functional studies have revealed two new ciliary complexes, CPLANE and KIAA0753-OFD1-FOPNL, and better characterised the TZ and MKS module.
Summary of OFD phenotype as well as localisation and function of OFD proteins
At the centrosomal level, the positive regulator C2CD3 was found to be an antagonist of OFD1, a negative regulator of centriole elongation.6 KIAA0753 or OFIP (OFD1 and FOR20 Interacting Protein) forms a ternary complex with OFD1 and FOPNL (also known as FOR20) to initiate ciliogenesis and control centriole length.34 When KIAA0753 is necessary to recruit OFD1 and FOPNL in centriole and pericentriolar satellites and to stabilise microtubule organisation in the centrosome, C2CD3 was thought to be associated with the KIAA0753-OFD1-FOPNL complex probably via OFD1 protein. Knockdown of OFD1, C2CD3 or KIAA0753 induces hyperelongated (OFD1 and KIAA0753) or shortened centrioles with the absence of subdistal appendages (C2CD3). These centriole defects affect membrane anchoring with the absence of cilia or greatly decreased cilium length. All these proteins control centriole elongation as do other centrosomal complexes, consisting of subunits with antagonist functions in ciliogenesis.
At the basal body level, a new protein complex, CPLANE (Ciliogenesis and Planar polarity Effectors) formed by FUZ, RSG1 and the three OFD proteins INTU, WDPCP and C5orf42, was characterised.33 C5orf42 initially recruits CPLANE components in the hierarchical assembly of this complex. CPLANE complex binds extensively with the IFT-A complex involved in retrograde intraflagellar transport, which is crucial for the recruitment of peripheral IFT-A proteins (IFT144, IFT43, IFT121 and IFT139) and their cytosolic preassembly. CPLANE defects affect intraflagellar transport and induce shortened cilia. Thus, RSG1 and FUZ genes are good candidates for OFD syndrome but, so far, Sanger sequencing of a local cohort negative for known OFD genes has not revealed any mutations in these genes.
At the TZ, two functional modules, MKS and NPHP, interact to regulate ciliogenesis, the assembly of membrane-microtubule Y-link connectors, diffusion barrier formation and the entry of IFT particles into the cilia.30 31 49 The NPHP module consists of two subunits (NPHP1–4) and the MKS module of twelve subunits (RPGRIP1L, TMEM107, TMEM216, B9D1, B9D2, MKS1, TMEM17, TMEM231, TMEM218, TMEM237, TMEM67 and CC2D2A), some of which are now known to be involved in OFD syndromes (TMEM231 and TMEM216). It has been reported that TMEM107 occupies a new intermediate layer in the hierarchical assembly of the MKS module and is necessary to recruit TZ proteins MKS1, TMEM17, TMEM237 and the new OFD protein TMEM231.30 In Caenorhabditis elegans, CEP290 is required for the TZ localisation of the MKS protein module and of other TZ proteins, such as TMEM138, involved in OFD syndrome.31
The new IFT57 gene encodes a peripheral subunit of the IFT-B complex, which consists of 14 members. It is believed that the IFT-B complex has been highly conserved during evolution and has an essential function in the formation and maintenance of primary cilia. Only five subunits are involved in ciliopathies (IFT27, IFT80, IFT81, IFT88 and IFT172).50 IFT57 mutations induce staining of IFT57 in the basal body in OFD patients’ fibroblasts, whereas IFT57 was observed in the whole cilia in controls. Likewise, the IFT57 mutation affects the Sonic HedgeHog pathway, thus confirming the involvement of IFT57 in ciliary transport and signalling transduction.32
Most of the genes involved in the same ciliopathy encode for subunits of the same protein complex and usually affect one ciliary function. In contrast, OFD syndromes implicate several protein complexes with various localisations and various ciliary functions, from centriole elongation to intraflagellar transport, thus illustrating the wide genetic heterogeneity. However, we noted a correlation between the genotype and the phenotype. Mutations in TZ genes mainly caused OFDVI, CPLANE-gene mutations caused unclassified OFD with cardiac malformations and mutations in genes coding for centrosomal proteins were implicated in various subtypes (OFDI, IX, XIV or unclassified) but with a clinical continuum between C2CD3, KIAA0753 and OFD1, sometimes including the MTS on brain MRI or renal abnormalities.
OFD syndromes: a distinct subgroup of ciliopathies and phenotype–genotype correlations
OFD syndromes were initially classified as 13 clinical subtypes depending on the additional clinical features (polycystic kidney disease, corpus callosum agenesis, tibial dysplasia, retinopathy and others). While numerous cases of OFDI, OFDIV and OFDVI syndromes have been reported, only anecdotal or single cases of some other subtypes have been published. This initial classification now appears to be obsolete given the wide clinical and molecular heterogeneity, with different overlapping ciliopathies such as JBS, MKS, BBS, SRP and NPHP. When OFD1 mutations induce OFDI or OFDVI subtypes, the OFDVI subtype appears to be linked to different genes also implicated in JBS and MKS. Considering the clinical and molecular data, the OFD classification could be reduced to three main subtypes and several additional anecdotal cases (table 3). Indeed, while a fine clinical description of the disease remains important for reverse phenotyping, prognosis and genetic counselling, a detailed classification appears to be extremely complex and of little use in such diseases with high clinical and genetic heterogeneity. Indeed, this high genetic heterogeneity leads to the use of WES for the molecular diagnosis of patients with OFD syndromes.
Novel classification of oral–facial–digital (OFD) syndrome based on associated clinical feature and molecular basis. MTS, molar tooth sign.
The high efficiency of the WES strategy in highly heterogeneous diseases
Despite the high clinical and genetic heterogeneity of these diseases, the solo WES strategy was very successful and led to the identification of five new genes responsible for OFD (C2CD3, KIAA0753/OFIP, IFT57, INTU and TMEM107). It also confirmed that OFD, BBS, JBS, MKS and SRP are allelic disorders and extended the clinical spectrum of TMEM138, TMEM231, C5orf42, C2CD3 and WDPCP genes, thus increasing to 16 the number of genes known to be responsible for OFDS (figures 3 and 4). This was possible thanks to a large 15-year international cohort and to knowing the probable mode of inheritance and the functions of candidate proteins. However, 42% of affected cases remained negative, raising questions about the choice of strategy. Indeed, the hypothesis of autosomal recessive inheritance and the limited availability of parental DNA at the beginning of the study led us to preferentially use the solo WES strategy, which is known to be less effective for the identification of sporadic mutations. In these negative patients, a trio WES strategy or whole-genome sequencing (WGS) could now be considered to look for non-exonic variants. In these negative cases, genetic counselling remains difficult because an autosomal recessive mode of inheritance could be excluded.
In conclusion, this solo WES strategy in 24 cases with OFDS identified five new genes responsible for OFD (C2CD3, KIAA0753/OFIP, IFT57, INTU and TMEM107) confirmed that OFD, BBS, JBS, MKS and SRP are allelic disorders and extended the clinical spectrum of TMEM138, TMEM231, C5orf42, C2CD3 and WDPCP genes, thus increasing to 16 the number of genes known to be responsible for OFDS (figures 3 and 4). Negative patients explored by secondary WES or WGS analysis with the trio strategy could extend these results to additional new genes.
Acknowledgments
We thank the patients and their families for their participation. We thank the Integragen Society, Centre National de Génotypage and Institute of Genetics and Molecular and Cellular Miology for the exome analyses and the Ferdinand Cabanne Biological Resources Centre for access to the biobank. We also thank the National Heart, Lung and Blood Institute GO Exome Sequencing Project (see URLs) and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women's Healt Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
References
Footnotes
Contributor BF, DD, RHG, CAJ, LB, MM, ID, GP, BD, BG-D, BR, ES-G, CB, IP, AF-E, AD, AD, AG, EB, DG, JA, DB, SRP, VCD, GJP, VH-P, LP, PL, SS, AM, TA-B, LF and CT-R ascertained the family and delineated OFD syndromes.
A-LB, JT, LJ, EL, MAH, VC, BA, NG, JSO, TE, JSL, OR, MRL, JBW, OEB, MVN, JBR performed molecular analysis, interpretation of results in these families and characterised ciliopathy proteins.
YD, J-FD, J-BR, A-LB performed the bioinformatic analysis of the data.
All the authors participated to the writing and reviewing processes of the manuscript.
Funding This work was supported by grants from the GIS-Institut des Maladies Rares (HTS), the French Foundation for Rare Diseases, the French Ministry of Health (PHRC national 2010-A01 014–35 and 2013) and the Regional Council of Burgundy.
Competing interests None declared.
Patient consent Pictures of patients described in this publication have previously been published.
Provenance and peer review Not commissioned; externally peer reviewed.