Article Text

Abstract
Background The effect of complex alleles in cystic fibrosis (CF) is poorly defined for the lack of functional studies.
Objectives To describe the genotype–phenotype correlation and the results of either in vitro and ex vivo studies performed on nasal epithelial cells (NEC) in a cohort of patients with CF carrying cystic fibrosis transmembrane conductance regulator (CFTR) complex alleles.
Methods We studied 70 homozygous, compound heterozygous or heterozygous for CFTR mutations: p.[Arg74Trp;Val201Met;Asp1270Asn], n=8; p.[Ile148Thr;Ile1023_Val1024del], n=5; p.[Arg117Leu;Leu997Phe], n=6; c.[1210-34TG[12];1210-12T[5];2930C>T], n=3; p.[Arg74Trp;Asp1270Asn], n=4; p.Asp1270Asn, n=2; p.Ile148Thr, n=6; p.Leu997Phe, n=36. In 39 patients, we analysed the CFTR gating activity on NEC in comparison with patients with CF (n=8) and carriers (n=4). Finally, we analysed in vitro the p.[Arg74Trp;Val201Met;Asp1270Asn] complex allele.
Results The p.[Ile148Thr;Ile1023_Val1024del] caused severe CF in five compound heterozygous with a class I–II mutation. Their CFTR activity on NEC was comparable with patients with two class I–II mutations (mean 7.3% vs 6.9%). The p.[Arg74Trp;Asp1270Asn] and the p.Asp1270Asn have scarce functional effects, while p.[Arg74Trp;Val201Met;Asp1270Asn] caused mild CF in four of five subjects carrying a class I–II mutation in trans, or CFTR-related disorders (CFTR-RD) in three having in trans a class IV–V mutation. The p.[Arg74Trp;Val201Met;Asp1270Asn] causes significantly (p<0.001) higher CFTR activity compared with compound heterozygous for class I–II mutations. Furthermore, five of six compounds heterozygous with the p.[Arg117Leu;Leu997Phe] had mild CF, whereas the p.Leu997Phe, in trans with a class I–II CFTR mutation, caused CFTR-RD or a healthy status (CFTR activity: 21.3–36.9%). Finally, compounds heterozygous for the c.[1210-34TG[12];1210-12T[5];2930C>T] and a class I–II mutation had mild CF or CFTR-RD (gating activity: 18.5–19.0%).
Conclusions The effect of complex alleles partially depends on the mutation in trans. Although larger studies are necessary, the CFTR activity on NEC is a rapid contributory tool to classify patients with CFTR dysfunction.
- nasal brushing
- [R74W;V201M;D1270N]
- [I148T;3199del6bp]
- [L997F;R117L]
- gating activity
Statistics from Altmetric.com
Introduction
Cystic fibrosis (CF) is a multisystem disease caused by mutations causing deficient or dysfunctional CF transmembrane conductance regulator (CFTR) protein. Typically, CF is characterised by elevated sweat chloride levels (SCL), obstructive lung disease, chronic bacterial infections of lower airways and sinuses, bronchiectasis and male infertility due to obstructive azoospermia. Most patients with CF have pancreatic insufficiency (PI) although 10–15% have normal exocrine pancreatic function and frequently show a milder clinical picture. Often, such cases have at least one CFTR mutation with a mild functional effect.1
Moreover, an increasing number of patients is diagnosed as CFTR-related disorders (CFTR-RD)2 generally characterised by a later onset of symptoms involving a single organ (ie, pancreatitis, disseminated bronchiectasis, obstructive azoospermia secondary to congenital bilateral absence of vas deferens (CBAVD)),3 usually associated to borderline SCL and mutations causing a different degree of CFTR protein dysfunction either in coding4–6 and in non-coding CFTR gene regions.7 ,8 Finally, the growing proportion of newborn screening (NBS) programmes revealed a large number of infants having a Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID) including those infants with discordance between immunoreactive trypsinogen at the NBS, SCL, CFTR genotype and clinical phenotype.9 ,10
More than 2000 CFTR mutations have been recorded so far worldwide (http://www.genet.sickkids.on.ca/app) although only a small number of them are clearly defined as CF-causing on the basis of functional studies as reported in the CFTR2 site (http://www.cftr2.org/index.php). Indeed, the use of CFTR gene sequencing11 leads frequently to the detection of mutations for which it lacks a clear and univocal genotype–phenotype correlation12–14 also because the genetic background and the environment in which each patient lives contribute to the individual CF phenotype.12 Furthermore, the existence of complex alleles complicates even more genetic counselling.15 ,16 They result from the combination of two or more CFTR mutations in cis (ie, on the same allele) that usually act as a pathogenic mutation whereas each single mutation has only a minor or none effect. Few subjects bearing the p.[Arg74Trp;Val201Met;Asp1270Asn],14 ,16 ,17 the p.[Ile148Thr;Ile1023_Val1024del],18 ,19 the p.[Arg117Leu;Leu997Phe]20–22 and the c.[1210-34TG[12];1210-12T[5];2930C>T]23 complex alleles were described so far with variable characteristics. However, a functional characterisation of the effect of these mutations was performed in a limited number of cases.17 ,23
We studied a cohort of patients with CF carrying CFTR complex alleles and described the genotype–phenotype correlation and the results of either in vitro and ex vivo studies performed on nasal epithelial cells (NEC).
Methods
Subjects population
The study was approved by the ethical committee of the University of Naples Federico II. We performed a retrospective analysis of all patients in follow-up at nine Italian CF centres and included all subjects who were homozygous or compound heterozygous for the following complex alleles: (1) p.[Arg74Trp;Val201Met;Asp1270Asn], n=8; (2) p.[Arg74Trp;Asp1270Asn], n=2; (3) p.[Ile148Thr;Ile1023_Val1024del], n=5; (4) p.[Arg117Leu;Leu997Phe], n=6; (5) c.[1210-34TG[12];1210-12T[5];2930C>T], n=3. Furthermore, we studied subjects homozygous or compound heterozygous for the following mutations: (1) (2) p.Asp1270Asn, n=2; (3) p.Ile148Thr, n=4; and (4) p.Leu997Phe, n=34. Finally, we studied obligate carriers heterozygous for the p.Ile148Thr (n=2); p.Leu997Phe (n=2) mutations and for the p.[Arg74Trp;Asp1270Asn] complex allele (n=2). We measured the CFTR gating activity on NEC from 39 subjects and compared the data with those obtained from: (1) patients with CF with two class I–II mutations, n=8, and (2) carriers of class I–II mutations, n=4.
Clinical data
The diagnosis of CF and CFTR-RD was performed according to the published criteria.2 ,24 CFSPID subjects were defined as above reported.9
For each subject, we collected a database including demographic, clinical and genetic data at diagnosis and during the follow-up. SCL were obtained by the Gibson and Cooke method.25 ,26 It was tested at diagnosis and repeated at the current age in patients with CFTR-RD or CFSPID (then, we considered the last available value) and twice at diagnosis in patients with CF. In each centre, sweat test was always performed in the same laboratory, thus ruling out the lack of harmonisation between different labs27 and all of them participate in the National Quality Control Programme.28 SCL were considered normal for values <40 mmol/L, borderline between 40 and 59 mmol/L and pathological ≥60 mmol/L.26 The last best forced expiratory volume in 1 second (FEV1), expressed as percentage of predicted value for age, according to standardised reference equations for spirometry29 and performed when patient was free from pulmonary exacerbations, was recorded for patients aged over 6 years. Given the interindividual variability of FEV1 and the evolution of lung damage with age, the patients were classified as severe or mild according to Schluchter et al30 criteria that take into account both FEV1 value and age. For patients who had died, we considered the last available value. Pseudomonas aeruginosa chronic infection was defined according to the modified Leeds criteria.31 Pancreatic sufficiency (PS) was defined on the basis of at least two values of faecal pancreatic elastase higher than 200 μg/g measured outside acute gastrointestinal diseases.32 Faecal pancreatic elastase was evaluated annually in patients with PS and at least 3 months before enrolment. Pancreatitis was defined according to the report from the international study group of paediatric pancreatitis.33 CF-related diabetes (CFRD) was diagnosed according to the American Diabetes Association criteria.34 CF-associated liver disease was defined by clinical and/or biochemical abnormalities when imaging demonstrated hepatic parenchymal abnormalities and/or portal hypertension in the absence of other demonstrated causes of liver disease.35 Complications such as allergic bronchopulmonary aspergillosis,36 meconium ileus (MI) or distal intestinal obstruction syndrome (DIOS), nasal polyposis, haemoptysis, and pneumothorax were also registered.
Molecular analysis of CFTR
We screened for a commercial panel of mutations with a detection rate of about 80%37; then, we looked for the most common rearrangements38 and carried out gene sequencing (detection rate about 97% for classic CF)39 in cases where one or both mutations resulted undetected after first-level analysis, according to European recommendations.11 Furthermore, we analysed intragenic CFTR short tandem repeats (STR) to exclude the recurrent origin of several CFTR mutations.40 All laboratories involved in this study take part in the national project on standardisation and quality assurance for molecular genetic testing.41 For CFTR mutations, we used the nomenclature guidelines suggested by the Human Genome Variation Society. However, for each mutation studied, online supplementary table 1 reports also the legacy name.
supplementary table
CFTR mutation nomenclature (HG Variation Society guidelines and Legacy name)
Nasal brushing and culture of NEC
NEC were collected by nasal brushing in 39 subjects. Details are provided as online supplementary materials.
Quantitative analysis of CFTR channel activity on NEC
Reported as online supplementary material.
supplementary material
HEK293 cell culture
Reported as online supplementary material.
Plasmid constructs and lentiviral vector production
Reported as online supplementary materials.
Western blot analysis
Reported as online supplementary material.
CFTR activity assay on HEK293 cells
Reported as online supplementary material.
Statistics
The t-test was used to compare the levels of CFTR gating activity on NEC between different groups.
Results
p.[Arg74Trp;Val201Met;Asp1270Asn] and p.[Arg74Trp;Asp1270Asn] complex alleles and p.Asp1270Asn mutation
Eight subjects were compound heterozygous for the p.[Arg74Trp;Val201Met;Asp1270Asn] complex allele (table 1).
Demographic and clinical data of subjects bearing the [p.Arg74Trp;p.Val201Met;p.Asp1270Asn] or the [p.Arg74Trp;p.Asp1270Asn] complex alleles or the p.Asp1270Asn mutation
Six of eight had a class I–II mutation in trans (ie, p.Phe508del: three cases; p.Asn1303Lys: two cases and p.Ser1206*: one case) and two of eight had in trans a CFTR mutation with higher residual function (ie, p.Asp1152His and p.Asp579Gly). Among patients compound heterozygous with a class I–II mutation, four of six were diagnosed with CF. These four patients with CF had pathological SCL, PS, mild lung disease and none was colonised by P. aeruginosa; two patients had impaired glucose tolerance (IGT). The mean CFTR gating activity on NEC (available from three patients with CF) was 11.2% (range 9.8–12.0%), significantly higher (p<0.001) as compared with the mean activity of 6.9% found in eight patients with two class I–II mutations (figure 1 and table 2).
CFTR gating activity measured on epithelial nasal cells in the following groups of patients: (A) CF with PI and two class I–II CFTR mutations; (B) CF with PI compound heterozygous for the [p.Ile148Thr;p.Ile1023_Val1024del] complex allele and a class I–II CFTR mutation; (C) CF with PS and compound heterozygous for a complex allele and a class I–II CFTR mutation; (D) CFTR-related disorders; (E) healthy subjects compound heterozygous for a class I–II mutation and a sequence variation with no functional effect; (F) healthy subjects heterozygous for a class I–II mutation; (G) healthy subjects heterozygous for a sequence variation with no functional effect; and (H) subjects with a undefined diagnosis
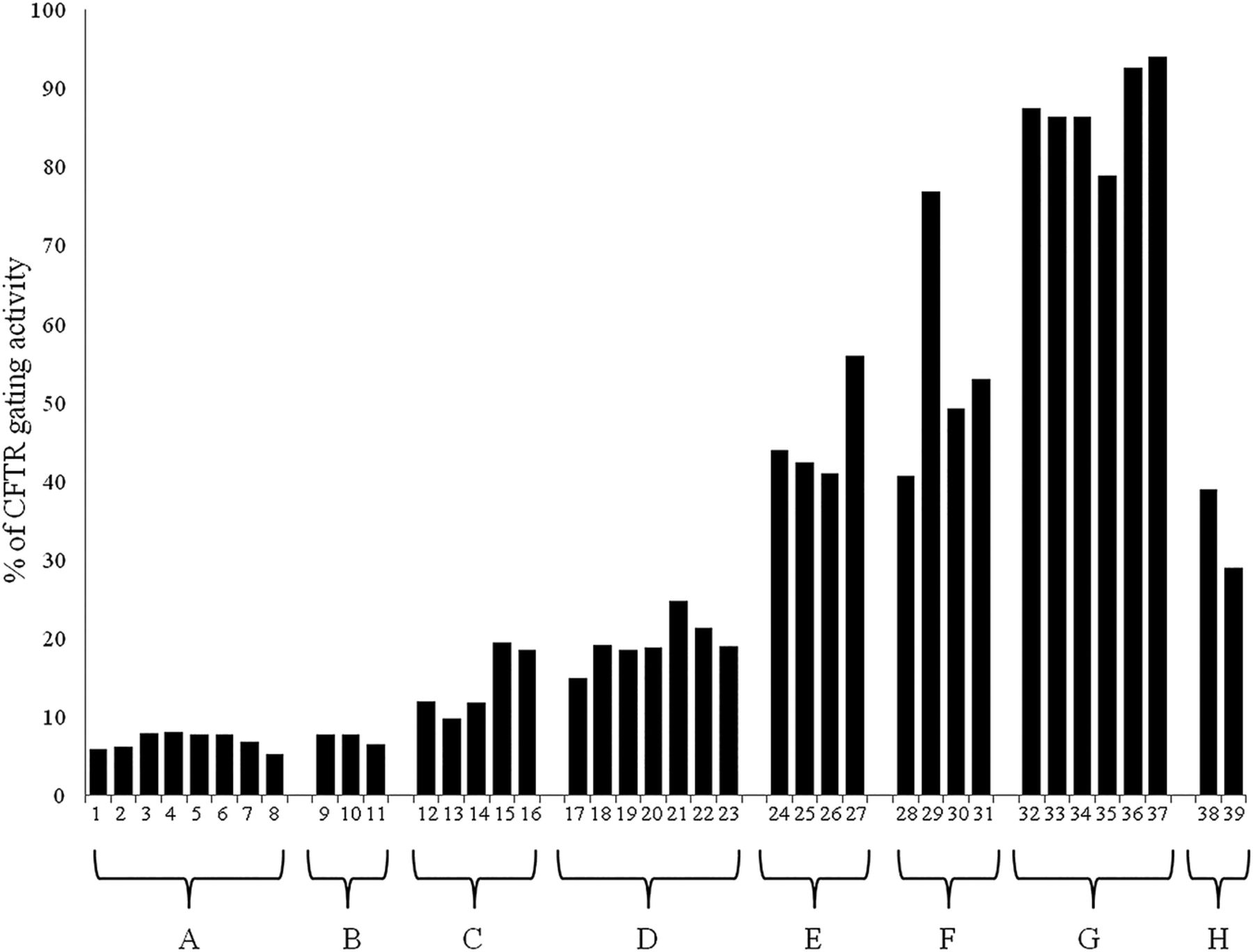
Cystic fibrosis transmembrane conductance regulator (CFTR) gating activity measured on epithelial nasal cells in several groups of subjects. The values obtained for each sample and the groups are reported in table 2.
The two other patients (one with the p.Phe508del mutation and the other with the p.Ser1206* mutation in trans), aged 12 and 13 years, were diagnosed as CFTR-RD despite normal SCL (ie, 37 mmol/L) in one of them. The CFTR gating activity on NEC was available only for the first patient and revealed a residual activity of 15.0% (table 1).
The two remaining patients with the p.[Arg74Trp;Val201Met;Asp1270Asn] complex allele, carrying the p.Asp579Gly and the p.Asp1152His mutation in trans, respectively, were identified because CBAVD at the age of 37 and 48 years, respectively. The patient carrying the p.Asp579Gly mutation had altered SCL (ie, 118 mmol/L) despite the monosymptomatic clinical course and a residual CFTR activity on NEC of 19.1%; the other subject, carrying the p.Asp1152His mutation, had normal SCL and CFTR gating activity of 18.5% (table 1).
Of the two subjects with the p.[Arg74Trp;Asp1270Asn] complex allele, one was classified as CFTR-RD and had the c.[1210-34TG;12 1210-12T[5]] complex allele in trans. He had CBAVD alone, normal SCL and a CFTR gating activity on NEC of 18.9% (table 1). The other, revealed by NBS, was previously diagnosed as CFSPID and now classified as healthy at the age of 5 years old.
Both clinical data and SCL were normal in the two subjects with the p.Asp1270Asn mutation (in trans with a class I–II mutation). The CFTR gating activity on NEC, available only for one of them, was 44.0% (figure 1 and table 2). Finally, we analysed the residual CFTR gating activity on NEC from two carriers of the p.[Arg74Trp;Asp1270Asn] complex allele. They had values of 92.6% and 94.0%, respectively.
Furthermore, we studied in vitro either the synthesis of CFTR protein by western blot and the CFTR gating activity in HEK293 cells transfected with the different mutations of the p.[Arg74Trp;Val201Met;Asp1270Asn] complex allele. Western blot analysis revealed two bands (figure 2). The C band represents the mature, fully glycosylated protein, while the B band represents the core-glycosylated protein. We calculated, for each mutant, the ratio between the C band and the total protein (band B+C). For the p.[Arg74Trp;Val201Met;Asp1270Asn] mutant, we obtained a ratio of 21%; the p.[Arg74Trp;Asp1270Asn] double mutant gave a ratio of 64% and, finally, the p.Asp1270Asn mutant is associated with a ratio of 83% (figure 2). These data compare with a 58% and 40% ratio obtained for the p.Phe508del and for the p.Asn1303Lys mutants, respectively (figure 2). We then evaluated the CFTR activity, that is, the rate of iodide efflux (figure 3) that was as high as 38.6%, 42.8% and 45.4% of the wild type for the triple, the double and the single mutant, respectively. These data compare with the values of 2.9% and 0.2% obtained for the p.Phe508del and for the p.Asn1303Lys mutants, respectively.

Western blot analysis of the cystic fibrosis transmembrane conductance regulator (CFTR) protein glycosylation in HEK293 cells stably expressing the wild type (1) or the mutant p.Phe508del (2), p.Asn1303Lys (3), p.[Arg74Trp;Val201Met;Asp1270Asn] (4), p.[Arg74Trp;Asp1270Asn] (5) and p.Asp1270Asn (6) proteins. Band C represents the mature, fully glycosylated protein, whereas band B represents the unglycosylated protein. The histogram shows the C/B+C ratio. The values are 1:98%, 2:58%, 3:4%, 4:21%, 5:65% and 6:86%.

{kind=link}
{kind=link}
{kind=link}
(A) Changes of fluorescence of stimulated HEK293 cells stably expressing the wild type (wt) or the mutants p.Phe508del, p.Asn1303Lys, p.[Arg74Trp;Val201Met;Asp1270Asn] (RDV), p.[Arg74Trp;Asp1270Asn] (RD) and p.Asp1270Asn (D) CFTR protein (mixture of 20 mM forskolin and 100 mM IBMX). The values were expressed as relative fluorescence F/F0, where F is the change in fluorescence with time and F0 is the minimum fluorescence. (B) The rate of fluorescence change was quantified from the maximal slope using the best fitting of the fluorescence change and was (1) wt: 100%, (2) p.Phe508del: 2.9%, (3) p.Asn1303Lys: 0.2%, (4) RDV: 38.6%, (5) DV: 42.8% and (6) D: 45.4%.
p.[Ile148Thr;Ile1023_Val1024del] complex allele and p.Ile148Thr mutation
All the five patients with the p.[Ile148Thr;Ile1023_Val1024del] complex allele (table 3) had a class I–II mutation in trans (ie, p.Phe508del: two subjects; p.Lys684SerfsX38: one subject; p.Asn1303Lys: one subject and p.Gly85Glu: one subject).
Demographic and clinical data of subjects bearing the [p.Ile148Thr;p.Ile1023_Val1024del] complex allele or the p.Ile148Thr mutation
All the patients had pathological SCL and PI; lung function ranged from normal (three cases) to severe (two cases) lung impairment. Three patients had CFRD and two had severe liver disease that was the cause of death (table 3). The mean CFTR gating activity (available only for three patients) on NEC was 7.3% (range 6.5–7.8%; figure 1 and table 2). Such a value was not significantly different as compared with the mean value of 6.9% obtained in patients with CF with two class I–II mutations.
All four subjects with the p.Ile148Thr mutation were compound heterozygotes with a class I–II mutation on the other allele. They were adults, asymptomatic and had normal SCL (table 3). They were revealed as CF carrier by molecular analysis, being consanguineous of patients with CF. The CFTR gating activity on NEC ranged from 41.0% to 56.0% (figure 1 and table 2). Finally, two further healthy subjects were revealed as heterozygous for the p.Ile148Thr mutation being partner of CF carriers. They had a CFTR activity on NEC of 87.4% and 86.3%, respectively.
p.[Arg117Leu;Leu997Phe] complex allele and p.Leu997Phe mutation
Two siblings were homozygous for the p.[Arg117Leu;Leu997Phe] complex allele (table 4, reported as online supplementary material). One is a woman diagnosed with CF with PS at 48 years because of recurrent pneumonia and chronic colonisation by P. aeruginosa. The CFTR gating activity on NEC was 39.0% (figure 1 and table 2). Her sibling is a 58-year-old male with CBAVD alone, and a SCL of 88 mmol/L. We also studied two pairs of siblings compound heterozygous for the p.[Arg117Leu;Leu997Phe] complex allele and the pArg334Trp (1 sib-pair) or the p.Gly85Glu (the other sib-pair, table 3) mutations. All subjects were affected by CF with PS. The CFTR gating activity on NEC, available only for one adult female (compound heterozygous for the p.Arg334Trp mutation), was 19.5% (figure 1 and table 2).
Demographic and clinical data of subjects bearing the [p.Arg117Leu;p.Leu997Phe] complex alleles or the p.Leu997Phe mutation
Moreover, we observed two subjects homozygous for the p.Leu997Phe mutation (table 4). The first has CBAVD alone and borderline SCL. The second, at the age of 21 years, has only chronic sinus disease with nasal polyposis (found at the age of 8 years old) and normal SCL. The CFTR gating activity on NEC was 28.9%.
Eight patients compound heterozygous for the p.Leu997Phe and a class I–II mutation and six patients compound heterozygous for the p.Leu997Phe and another mutation (table 4) had monosymptomatic CFTR-RD (CBAVD: nine cases; recurrent pancreatitis: three cases; isolated bronchiectasis: two). Six of them had borderline SCL and eight had normal SCL. In two patients from this group, both carrying a class I–II mutation on the other allele, the CFTR residual gating activity on NEC was 24.8% and 21.3%, respectively.
Nine subjects (aged 2–5 years old) compound heterozygous for the p.Leu997Phe mutation and a class I–II mutation (four cases) or another mutation (five cases) had been classified as CFSPID. At present, all of them are asymptomatic (table 4) and have normal SCL.
Nine subjects (aged 31–46 years old) compound heterozygous for the p.Leu997Phe and a class I–II mutation (four cases) or another mutation (five cases) were classified as healthy, being all asymptomatic. Seven of the nine subjects had normal SCL, in two of the nine the SCL were in the borderline range over the last 2 years (60 mmol/L in both the cases in the last evaluation). The nine subjects had been identified for familiarity with patients with CF (six cases) or being partner of CF carrier subjects (three cases). For one of them, the CFTR activity on NEC was 36.9%.
Finally, two healthy subjects heterozygous for the p.Leu997Phe mutation were analysed for the CFTR gating activity on NEC that revealed values of 86.4% and 78.9% (tables 2 and 4, and figure 1).
In 10 subjects bearing the p.Leu997Phe mutation (including the two homozygous subjects), the analysis of the STR revealed that the IVS8CA, IVS17b TA and CA haplotype associated with the p.Leu997Phe mutation was invariably 20, 13 and 7, respectively, thus excluding a recurrent origin of the mutation.
c.[1210-34TG;[12];1210-12T[5]2930C>T] complex allele
We studied three patients with the c.[1210-34TG[12];1210-12T[5];2930C>T] complex allele in trans with a class I–II mutation (table 5).
Demographic and clinical data of subjects bearing the c.[1210-34TG[12];1210-12T[5];2930C>T] complex allele
One had CF with PS, a well-maintained pulmonary function despite P. aeruginosa chronic colonisation and pathologic SCL. Two other patients had CFTR-RD. The CFTR gating activity measured on NEC was, respectively, 18.5% in the patient with CF and 19.0% in the first of the two patients suffering for CFTR-RD (figure 1 and table 2).
Finally, supplementary table 2.
supplementary table
Synopsis of CF phenotypic expression in patients bearing different CFTR genotypes. CFTR-RD: CFTR related disorders; PS: pancreatic sufficiency; PI: pancreatic insufficiency
Discussion
This is the first study including a large number of subjects, carrying different CFTR complex alleles that were evaluated for the genotype–phenotype correlation and were analysed for the CFTR residual gating activity using the ex vivo system of NEC obtained by brushing.
The p.[Arg74Trp;Val201Met;Asp1270Asn] was found in eight patients and is a mild CF-causing mutation whose clinical impact is influenced by the mutation in trans. We found this mutation in trans with a class I–II mutation, resulting either in CF-PS (four patients) or in CFTR-RD (two patients). In two other patients, the mutation was in trans with mutations with intrinsic residual CFTR function (ie, p.Asp579Gly and p.Asp1152His) that usually have a less severe clinical impact.42 ,43 They had CBAVD, and they compare with four patients previously described with the same complex allele in trans with mild mutations and CBAVD alone.16 ,17 The mean CFTR gating activity on NEC in three patients with CF that had in trans a class I–II mutation resulted 11.2% versus a mean of 6.2% (p<0.001) found in patients with CF and two class I–II mutations and 17.5% in three patients with CFTR-RD, confirming the higher residual activity of the p.[Arg74Trp;Val201Met;Asp1270Asn] mutated protein. The study of in vitro expression (never previously performed for the triple mutant) was performed to define the effect of the complex allele without the influence of the mutation in trans. The p.[Arg74Trp;Val201Met;Asp1270Asn] construct causes a relevant reduction in the processing of the mature protein (ie, 21%), but it gives rise to a CFTR residual gating activity of 38% that compares with the residual activity of 2.9% and 0.2% observed for the p.Phe508del and for the p.Asn1303Lys mutants, respectively, adding on the milder functional effect of the p.[Arg74Trp;Val201Met;Asp1270Asn] complex allele as compared with class I–II mutations.
We found the p.[Arg74Trp;Asp1270Asn] complex allele in a child originally classified as CFSPID,9 ,44 currently asymptomatic at the age of 5 years old and in a patient with and normal SCL and CBAVD (with a residual CFTR activity on NEC of 18.9%). The p.[Arg74Trp;Asp1270Asn] complex allele has been reported only in two asymptomatic subjects so far.17 Furthermore, we observed two adults with the p.Asp1270Asn in trans with the p.Phe508del and the p.Asn1303Lys, respectively, both asymptomatic with normal SCL. So far, the p.Asp1270Asn sequence variation has never been described in subjects with CF or CFTR-RD and it was found with a high frequency in healthy subjects.17 The CFTR activity assessed on NEC from two heterozygotes for the p.[Arg74Trp;Asp1270Asn] complex allele revealed a residual gating activity >90% in both cases, and the activity obtained from a subject compound heterozygous for the p.Asp1270Asn and the p.Phe508del was 44.0%, comparable with that obtained from heterozygous carriers of the p.Phe508del mutations. Finally, either the [p.Arg74Trp;p.Asp1270Asn] complex allele or the p.Asp1270Asn mutation caused only a slight reduction in the synthesis of the mature protein (65% and 86%) in vitro and were associated with a gating activity of 43% and 45%, respectively. All these data (that however need to be confirmed on larger number of cases) indicate that such mutations are not enough to cause disease45 in contrast with the definition of CFTR2 that classifies the p.Asp1270Asn as a mutation with varying clinical consequence (http://www.cftr2.org/index.php).
The p.[Ile148Thr;Ile1023_Val1024del] complex allele acts as a severe CFTR mutation. In fact, it was observed in trans with a class I–II mutation in five patients with CF-PI and severe complications of the disease, and the CFTR gating activity on NEC was comparable with that observed in patients with CF with two class I–II mutations. On the contrary, the p.Ile148Thr variant does not have clinical impact: we found such sequence variation in trans with a class I–II CFTR mutation in four asymptomatic adults with a gating activity of CFTR on NEC ranging 41.0–56.0% (ie, in the range observed in carrier subjects of a class I–II mutation). Finally, the CFTR gating activity measured on NEC from two healthy volunteers heterozygous for the p.Ile148Thr (ie, 87.4% and 86.3%) further confirms that the sequence variation alone has a minimal functional effect, in agreement with the data obtained in model systems46 and with its high frequency in healthy subjects.17
We studied two siblings homozygous for the p.[Arg117Leu;Leu997Phe] complex allele (ie, the first cases with such a genotype at our knowledge), both diagnosed in adulthood. The first was diagnosed as CF for the altered SCL with a mild clinical course and the sibling had CBAVD alone with pathological SCL. Similarly, four other patients compound heterozygous for the complex allele and another CFTR mutation have mild CF with PS. The residual CFTR gating activity on NEC of 39.0% obtained in one of the homozygous patients and that of 19.5% obtained in one of the cases compound heterozygous for the complex allele and the p.Arg334Trp mutation indicates that the p.[Arg117Leu;Leu997Phe] mutation is associated with a higher residual function as compared with class I–II mutations. These data compare with four patients compound heterozygous for the p.[Arg117Leu;Leu997Phe] and another CFTR mutation that displayed a mild CF with PS in two cases and a more severe form of the disease with or without PI in two.20
We found the p.Leu997Phe homozygous or in trans with a known causing mutation either in patients with CFTR-RD (mainly CBAVD) or in healthy subjects, in agreement with previous reports.20 ,21 ,47 Furthermore, some of our subjects, classified as CFSPID in infancy, resulted free from symptom during the follow-up in the successive years, again in agreement with previous studies.48 Going to the functional analysis of CFTR on NEC, in two patients with CFTR-RD and both compound heterozygous for the p.Leu997Phe and a class I–II mutation (ie, the p.Gly542* and the p.Asn1303Lys, respectively), we obtained a residual activity of 21.3% and 24.8%, while the activity measured on an asymptomatic subject compound heterozygous with the p.[Phe508del];[p.Leu997Phe] genotype was 36.9%. Thus, the p.Leu997Phe is associated with a higher residual gating activity of CFTR as compared with class I–II mutations, but with a wide range of variability. The different clinical and functional impact of the p.Leu997Phe mutation does not appear to depend on the mutation in trans with the p.Leu997Phe. In fact, either among the CFTR-RD or among asymptomatic subjects, the p.Leu997Phe mutation was found in homozygosis or in trans with both class I–II mutations (ie, p.Phe508del, p.Asn1303Lys) and with mild mutations like the p.Asp1152His or the c.[1210-34TG[12];1210-12T[5]] complex allele.43 Furthermore, the STR analysis40 performed on several patients with CFTR-RD as well as on several asymptomatic subjects excluded the recurrent origin of the p.Leu997Phe mutation and, thus, the possibility that the mutation would be associated—in some cases—with other intragenic mutations that would interfere with its functional impact. We suggest that gene variants within the promoter region7 or at the 3′UTR8 may modulate CFTR expression explaining the variability of the p:Leu997Phe mutation. Of course, factors besides CFTR such as environment and modifier genes contribute to modulate the symptoms of the disease of each patient with CF.
Finally, the three patients with the c.[1210-34TG[12];1210-12T[5];2930C>T] complex allele had a severe mutation in trans, that is, p.Asn1303Lys: two cases and the c.579+1G>T: one case.49 One had a very mild clinical course and was classified as CF since SCL were 87 mmol/L. The remaining were classified as CFTR-RD. The CFTR gating activity was 18.5% (in the patient with CF) and 19.0% in one of the patients with CFTR-RD, indicating that the c.[1210-34TG[12];1210-12T[5];2930C>T] complex allele only partially impairs the CFTR activity and, thus, it acts as a mild mutation. These results (to be extended on a larger number of cases with the same genotype) are in agreement with those reported in a patient compound heterozygous for the c.[1210-34TG[12];1210-12T[5];2930C>T] complex allele and the p.Phe508del mutation with a mild course of the disease (similar to our three cases), even if nasal potentials and monocyte functional assay results were compatible with a CF phenotype despite borderline SCL.23
Some cases warrant a further comment due to the discordance between SCL (italics in tables 1⇑–3), symptoms and the residual CFTR gating activity on NEC. Two asymptomatic adults with the p.Leu997Phe (one compound heterozygous with the severe p.Phe508del and another with the mild c.[1210-34TG[12];1210-12T[5]]) had SCL of 60 mmol/L (confirmed during the last 2 years). Furthermore, some discordance between SCL and mild clinical symptoms was found in the two siblings homozygous for the p.[Arg117Leu;Leu997Phe] complex allele (the first of which had a residual CFTR gating activity as high as 39%). The high SCL observed in all these cases may depend on the strong functional effect that the p.Leu997Phe specifically exerts on the Cl− conductance in sweat cells observed in vitro.21 We found a discordance between SCL and the clinical expression of the disease also in patients with other genotypes, that is, the SCL of 118 mmol/L found in a patient compound heterozygous for the p.[Arg74Trp;Val201Met;Asp1270Asn] and the p.Asp579Gly mild mutation,42 again with a mild phenotype (CBAVD alone) and a residual gating activity not matching with the diagnosis of CF (ie, 19.1%). Finally, we found several patients with a clear clinical picture of CFTR-RD, a residual CFTR gating activity on NEC around 20% and SCL ranging 30–40 mmol/L that most authors report as normal values in adults26 even if the European recommendations for CFTR-RD diagnosis suggest that SCL between 30 and 40 mmol/L would be considered borderline.2 These discordances again point on the role of the whole genomic background of patients with CF and the environment in which they live in modulating the clinical expression of the disease. However, in most of such patients, the measure of the CFTR gating activity on NEC was in agreement with the clinical picture. This is a poorly invasive tool that may contribute (once validated on a higher number of cases) to classify the patients and may help to predict the clinical severity of the disease, particularly in cases where discordance between clinical picture, SCL and genetics would occur.
Acknowledgments
Grants from Fondazione per la ricerca sulla Fibrosi Cistica (Verona), project call 2013, are gratefully acknowledged.
References
Footnotes
Contributors VT, GC and FA conceived and designed the study, interpreted data and wrote the paper; DS and ML recruited patients and contributed to manuscript preparation; VR, AA, VC, NC, RC, CC, PI, VL, SQ, MS and VMS recruited patients; AMDL, AE, MC, MC, AP, RC and FZ analysed the CFTR gating activity on nasal epithelial cells and described the results of either in vitro and ex vivo studies. All authors approved the final manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethical committee of the University of Naples Federico II.
Provenance and peer review Not commissioned; externally peer reviewed.