Article Text

Abstract
Background A constellation of neurodegenerative disorders exists (Gordon Holmes syndrome, 4H leucodystrophy, Boucher-Neuhauser syndrome) in which patients suffer from both neurological disease (typically manifested by ataxia) and reproductive failure (idiopathic hypogonadotropic hypogonadism (IHH)). POLR3B, which encodes the second largest subunit of RNA polymerase III (pol III), and POLR3A, which forms the pol III catalytic centre, are associated with 4H leucodystrophy.
Methods Whole exome sequencing was performed on a large cohort of subjects with IHH (n=565). Detailed neuroendocrine studies were performed in some individuals within this cohort.
Results Four individuals (two of them siblings) were identified with two rare nucleotide variants in POLR3B. On initial evaluation, all subjects were free of neurological disease. One patient underwent treatment with exogenous pulsatile gonadotropin-releasing hormone for 8 weeks which failed to result in normalisation of his sex steroid milieu due to pituitary resistance.
Conclusions These findings suggest that the spectrum of phenotypes resulting from POLR3B mutations is wider than previously believed and that POLR3B can be associated exclusively with disorders characterised by abnormal gonadotropin secretion.
- POLR3B
- Ataxia
- Idiopathic Hypogonadotropic Hypogonadism
- 4H Leukodystrophy
- whole exome sequencing
Statistics from Altmetric.com
Introduction
Idiopathic hypogonadotropic hypogonadism (IHH) is a syndrome characterised by delayed or abnormal pubertal development and low sex steroids in the setting of low/normal gonadotropins. In the majority of patients, IHH is caused by a spectrum of abnormal gonadotropin-releasing hormone (GnRH) secretory patterns, including deficient synthesis or secretion of endogenous GnRH.1 Thus, the vast majority of patients can be successfully treated with administration of exogenous pulsatile GnRH, which stimulates the pituitary gland to produce gonadotropins, thereby restoring the endogenous production of sex steroids.2 In addition to the reproductive defects, some patients with IHH have additional somatic abnormalities (cleft lip, renal agenesis, digit abnormalities, etc).3 A rare subset of patients with IHH suffer from a severe neurodegenerative syndrome characterised by progressive ataxia, ±dementia, ±hypodontia, ±ocular abnormalities.
Appreciation for these complex neurodegenerative/reproductive syndromes began over a century ago when the association between cerebellar ataxia and hypogonadism was first described by Gordon Holmes.4 Biallelic inactivating mutations in RNF216 (encodes an E3 ligase), or the combination of deleterious mutations in RNF216 and OTUD4 (encodes a deubiquitinase) have been discovered as genetic causes of this syndrome.5 Boucher-Neuhäuser syndrome, first described in 1969, also incorporates ataxia and hypogonadism, but affected individuals have an additional clinical feature of chorioretinal degeneration.6 Boucher-Neuhäuser syndrome has recently been linked to mutations in PNPLA6 (encodes patatin-like phospholipase domain-containing protein-6, a phopholipase protein important to the central nervous system through neuron growth and retinal development).7 Mutations in PNPLA6 can cause both Gordon Holmes and Boucher-Neuhäuser syndromes, demonstrating the spectrum of neurodegenerative phenotypes that can be associated with mutations in a single gene.7
In addition to the Gordon Holmes and Boucher-Neuhäuser syndromes, autosomal recessive RNA polymerase III-related leucodystrophy (also called 4H leucodystrophy) has been described in which patients present with ataxia, hypogonadotropic hypogonadism and hypodontia.8 Mutations in POLR3A,9–11 which encodes subunit A of RNA polymerase III, POLR3B12 ,13 and most recently POLR1C,14 have been found to cause this disorder. POLR3B is the second largest subunit of Pol III and, together with POLR3A, forms the enzyme's catalytic centre.15 Similar to PNPLA6, mutations in these polymerase genes can lead to closely related neurological phenotypes including ataxia, delayed dentition and hypomyelination,16 hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum,17 tremor-ataxia with central hypomyelination,18 and leucodystrophy with oligodontia.19 In general, patients with POLR3B mutations have been found to have a slightly earlier onset of disease, but a less severe clinical course than those with mutations in POLR3A, which has been linked with faster regression and shorter life expectancy.20
Given that reproductive symptoms are often unappreciated in complex syndromic cases, we sought to investigate whether patients with hypogonadotropic hypogonadism—and no other neurological features—might carry mutations in POLR3B. We hypothesised that within a large cohort of patients with IHH and no neurological phenotype, we would identify individuals bearing mutations in this gene. We did indeed identify four such patients with IHH with two rare variants each in POLR3B. This discovery expands the complexity of the genetics of hypogonadal syndromes and suggests that genes underlying these complex neurodegenerative syndromes may also play a larger role in isolated hypogonadotropic states.
Methods
All activities were approved by the Partners Human Research Committee. All subjects provided written informed consent.
Patient cohort
Probands with IHH were referred to the Reproductive Endocrine Unit at Massachusetts General Hospital for participation in genetic studies. The diagnostic criteria for IHH included male or female individuals ≥18 years who lack sexual maturation or menses, with hypogonadotropic hypogonadism (T<100 ng/dL in men: E2<20 pg/mL in women) without any functional aetiology, a negative brain MRI (save for olfactory bulb abnormalities), otherwise normal anterior pituitary hormonal functions, and normal ferritin levels. Subjects can either have normosmic IHH or an impaired sense of smell (Kallmann syndrome (KS)=IHH+anosmia).21 No subject was known to have frank neurological abnormalities.
DNA sequencing
Five hundred and sixty-five individuals with IHH (±anosmia) underwent whole exome sequencing (WES) to understand the genetic pathomechanism underlying their condition. Genomic DNA was obtained from peripheral blood samples by standard phenol-chloroform extraction.
WES was performed using the Broad Institute's Illumina Capture platform using the HiSeq sequencer. All sequencing reads were aligned to the hg19 reference genome with Burrows-Wheeler alignment maximal exact matches (BWA-MEM).22 Variant annotation was performed with Variant Effect Predictor23 and the Genome Analysis Toolkit (GATK) Variant Annotator tool. Rare sequence variants were then identified based on a minor allele frequency <1% as determined by the ExAC (http://exac.broadinstitute.org) database with special consideration for the ethnicity of the patient. All sequence variations were confirmed in a separate PCR by direct sequencing. Genes and proteins are described using standard nomenclature by the Human Genome Variation Society (http://www.hgvs.org).
The severity of POLR3B variants was determined by previously published studies or prediction programs (PolyPhen,24 Mutation Taster25 and Panther,26 Mutation Taster, SIFT (Sorting Intolerant From Tolerant),27 PROVEAN (Provean Variation Effect Analyzer),28 pMut,29 and MutPred30 for non-synonymous changes, and NNSPLICE 0.9 version (http://www.fruitfly.org/seq_tools/splice.html) for splice site changes).
Neuroendocrine studies
Subject #1 was admitted to the Clinical Research Center of Massachusetts General Hospital for blood sampling every 10 min for 24 hours to assess Luteinizing hormone (LH) secretion.31 The subject then began treatment with exogenous pulsatile GnRH subcutaneously with frequent monitoring of his response. During his first inpatient visit to the Research Center, subject #1 received 5.0 ng/kg GnRH intravenous bolus (IVB) every 2 hours×8 hours. He was discharged on 5.0 ng/kg GnRH every 2 hours×2 weeks but through subcutaneous, not intravenous, administration. He was then readmitted to the research centre where he received 5.0 ng/kg GnRH IVB every 2 hours×6 hours. At the 6 hours time point, subject #1 was then transitioned to a higher GnRH dose (10.0 ng/kg GnRH IVB every 2 hours×6 hours). The patient was then discharged on 10.0 ng/kg GnRH subcutaneously every 2 hours×2 weeks. Thus, every 2 weeks subject #1's GnRH dose was sequentially increased (5–100 ng/kg) and his transition from one dose to the next was captured during inpatient studies. The LH responses were captured through blood sampling every 10 min. Samples were assayed for LH by Radioimmunoassay (RIA).32 LH pulses were assessed using a modification of the Santen and Bardin algorithm.33 ,34
Results
Of 565 IHH subjects, 4 were identified with two rare variants in POLR3B (figure 1). The histories of the latter four patients are presented below (table 1).
Clinical data of three patients screened for two rare variants in POLR3B
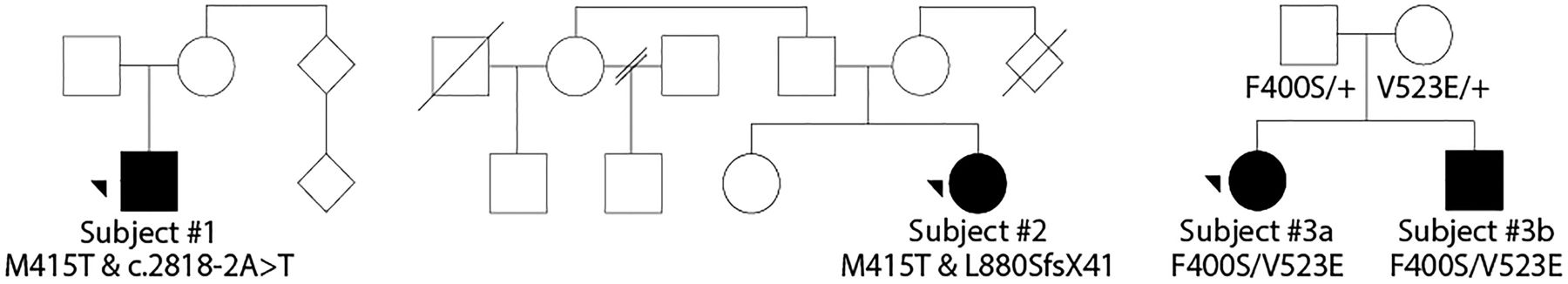
Pedigrees for subjects #1–3b. Affected individuals shaded in black.
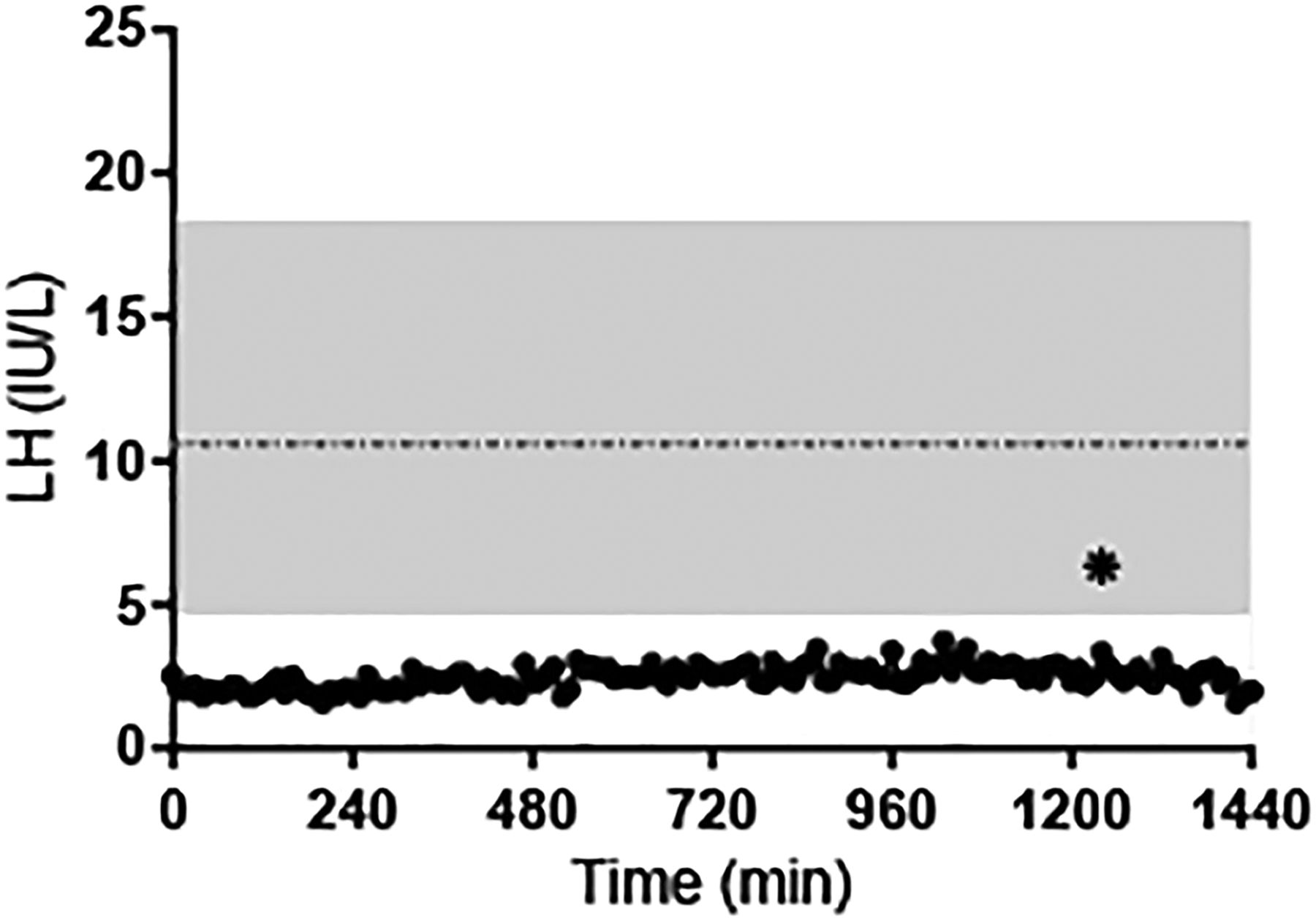
Subject #1 is a Caucasian man who presented to local providers at 14 years for delayed growth. He had scarce pubic and axillary hair, no facial hair and difficulty ejaculating. At age 21 years, the patient still had minimal pubic, axillary and facial hair, but his testicular volume was noted to be 15 cc. Laboratory studies revealed low gonadotropins with minimal increase after GnRH stimulation (LH: 2.3→4.5 mIU/mL, Follicle-stimulating hormone (FSH): 1.9→2.1 mlU/mL) and testosterone (T) of 170 ng/dL (figure 2). Note that the subject's LH concentrations were below the mean LH level (±2SD) found in 20 normal men, and below the lower limit of the normal range (4.7 mIU/mL) throughout the test.31 Stimulation testing revealed no anterior pituitary endocrinopathies. Subject #1 was treated with depotestosterone 100 mg intramuscularly every 3 weeks and continued to undergo diagnostic testing including a brain MRI which, at age 23 years, was reported as normal. He was also treated with clomid 50 mg twice daily for 7 days which resulted in no change in LH, FSH or T.
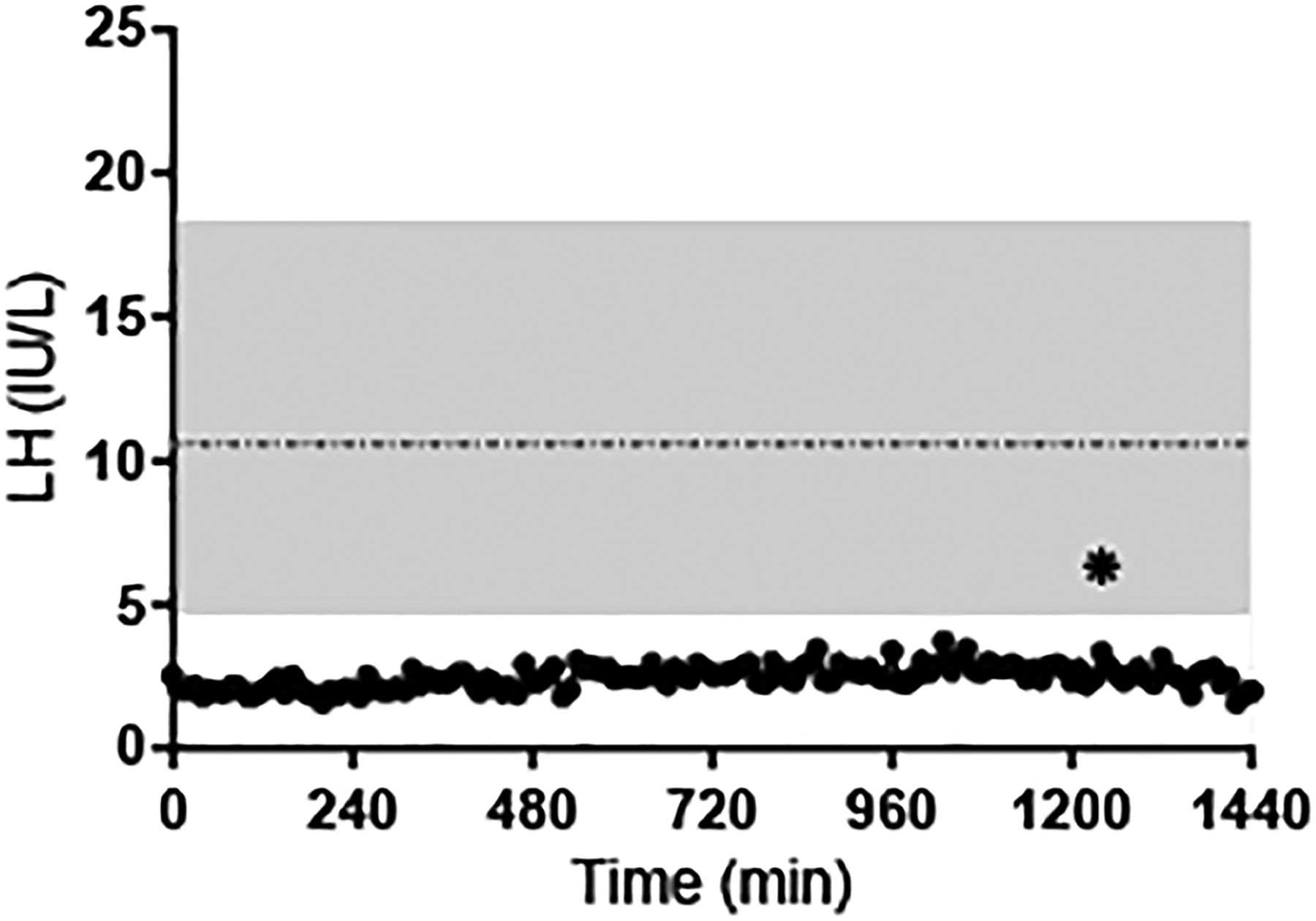
LH concentrations as determined by blood sampling every 10 min ×24 hours in subject #1 (28 years). Shaded area: range of mean LH values in 20 healthy men.31 Horizontal dashed line: mean LH value. *LH pulse as defined by a modified Santen and Bardin algorithm.
At 27 years, subject #1 presented to the Reproductive Endocrine Unit of Massachusetts General Hospital. Examination revealed Tanner 3 gynaecomastia, and testicular volume of 15 cc bilaterally. No neurological, dental or ocular anomalies were noted with the exception of horizontal nystagmus. Given his hypogonadotropism on laboratory studies but his robust testicular size on physical examination, subject #1 was felt to have the fertile eunuch variant of hypogonadotropic hypogonadism.
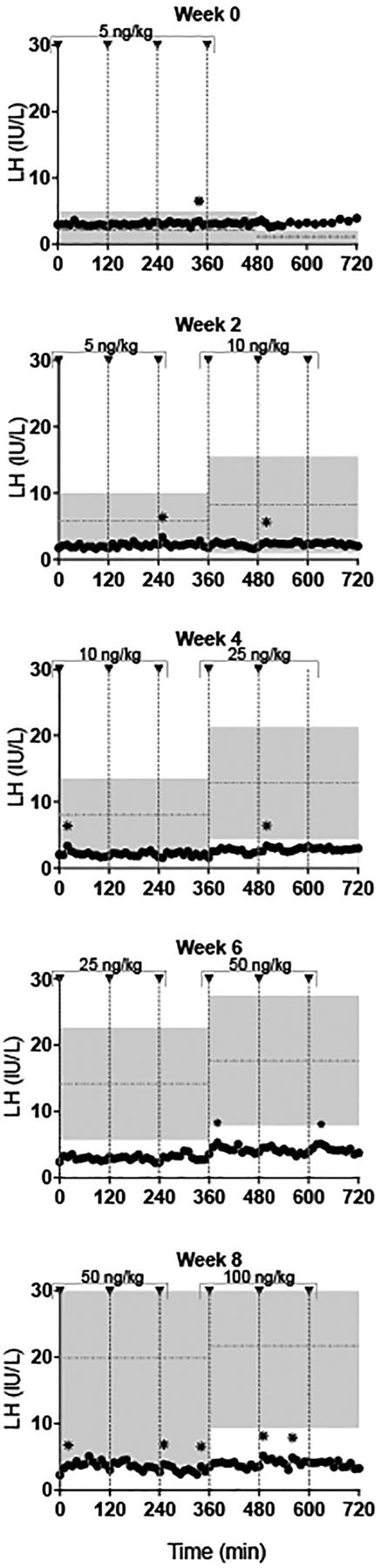
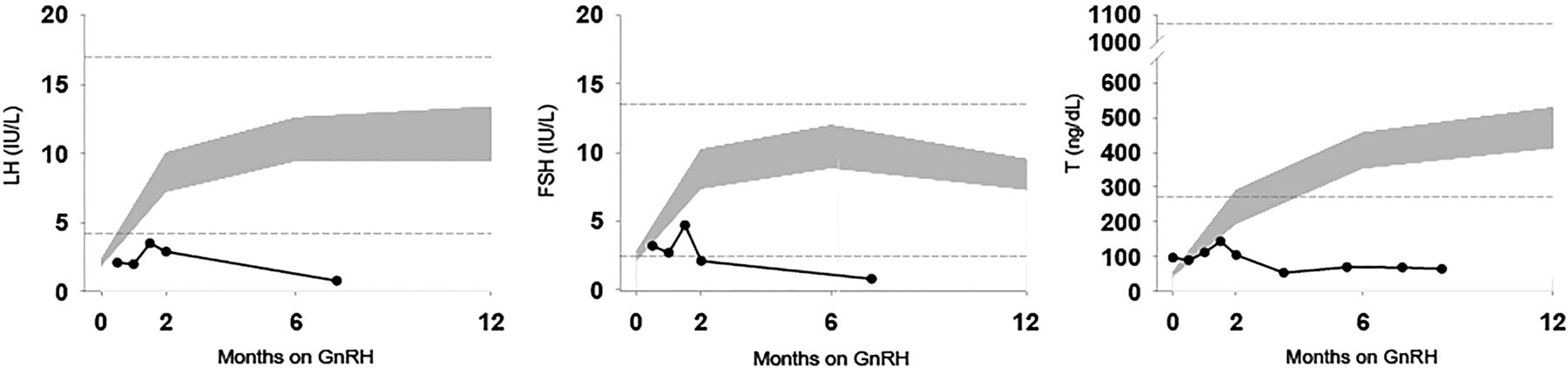
Subsequently, subject #1 initiated therapy with exogenous, pulsatile GnRH subcutaneously and intravenously (5.0 ng/kg escalating to100 ng/kg) over an 8 week interval (figure 3). He continued to receive escalating doses of GnRH (maximum dose 400 ng/kg) over an 8.5 month course. He never normalised his gonadotropin/testosterone levels like other male patients with IHH receiving identical therapy (figure 4).34 Eventually, subject #1 discontinued exogenous pulsatile GnRH subcutaneously and began human chorionic gonadotropin therapy subcutaneously.
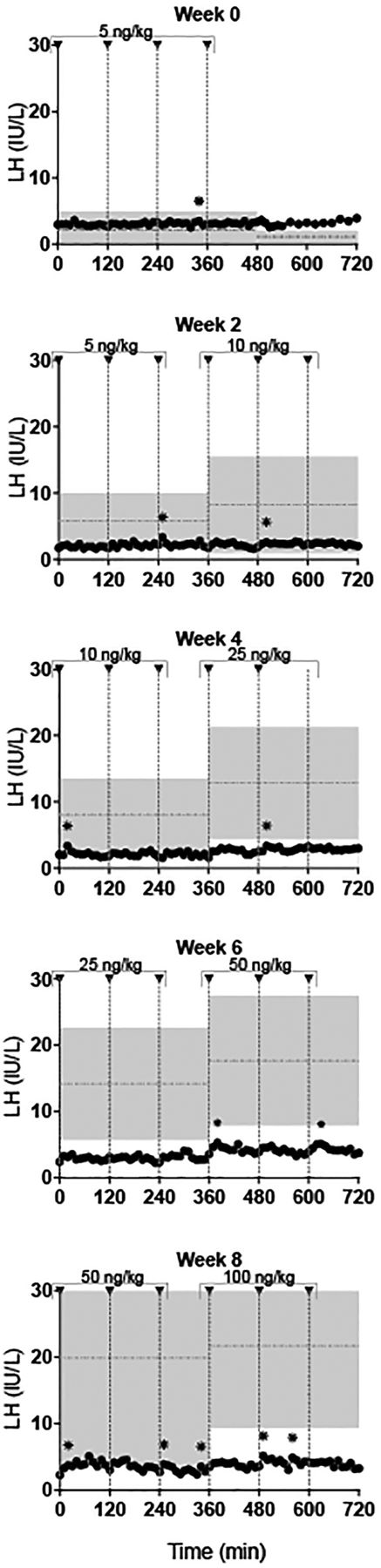
LH response of subject #1 to escalating doses of exogenous gonadotropin-releasing hormone (GnRH) IVB over an 8 week period. GnRH doses were sequentially increased (5–100 ng/kg, indicated by arrowheads) with transition from one dose to the next captured during inpatient studies. Shaded area: LH (mean±SD) in idiopathic hypogonadotropic hypogonadism men (n=12).34 Horizontal dashed lines: mean LH value.31 *LH pulse as defined by a modified Santen and Bardin algorithm.

Subject 1: LH, FSH and T levels in response to chronic therapy with exogenous pulsatile gonadotropin-releasing hormone (GnRH). Grey shaded area: 95% CI of GnRH-stimulated LH, FSH and testosterone in patients with idiopathic hypogonadotropic hypogonadism with a typical response to GnRH treatment.34 Horizontal dashed lines: upper and lower limits of serum hormone levels in healthy adult men.31
The results of subject #1's WES revealed a T-to-C transition in POLR3B leading to an amino acid change (c.[1244T>C] p.[Met415Thr]). He was also found to carry c.[2818A>T], which sits 2 bps outside of the splice site junction and, according to NNSPLICE (http://www.fruitfly.ord/seq_tools/splice.html), is predicted to create a splice defect.
Subject #2 is a Caucasian woman of mixed European descent who presented with amenorrhoea at 17 years. She had undergone a puberty related growth spurt in middle school, but experienced late development of her secondary sex characteristics. She had no axillary hair, and her breast development and pubic hair growth had stalled. Upon examination subject #2 did not show any neurological or dental anomalies; however she did have difficulty with spatial depth perception due to severe myopia. At 20 years she began hormone replacement therapies; however, after discontinuing these therapies her amenorrhoea returned. The subject's family history was notable for late menarche at 14 years in both her mother and sister, as well as exercise-induced hypothalamic amenorrhoea in her sister.
The results of subject #2's WES revealed a rare variant in POLR3B leading to an amino acid substitution (c.[1244T>C] p.[Met415Thr]) and a nucleotide insertion leading to a frameshift (c.[2633insT] p.[L880SfsX41]).
Subject #3a is a Caucasian woman who presented with primary amehorrhea at 18 years. She was diagnosed with hypodonadotropic hypogonadism and prescribed hormone replacement therapy. Upon her most recent evaluation at 31 years, she was reported to have had a normal neurological exam, during which she did not show any signs of neurological abnormalities, dental anomalies or ocular anomalies. Her brother, subject #3b carried a diagnosis of IHH and had a diminished IQ of 76, but no other neurological, dental or ocular abnormalities. The parents were unaffected with either reproductive or neurological disease. Subject #3b was treated with exogenous gonadotropins, which resulted in an improvement in the quality of his semen, an increase and normalisation of inhibin B, and an increase of testes to 12 cc bilaterally.
Subjects #3a and #3b both exhibited a heterozygous substitution in POLR3B (c.[1199T>C] p.[(Phe400Ser)]) (see figure 1; table 1) and a heterozygous missense change leading to a substitution (c.[1568T>A] p.[(Val523Glu)]).
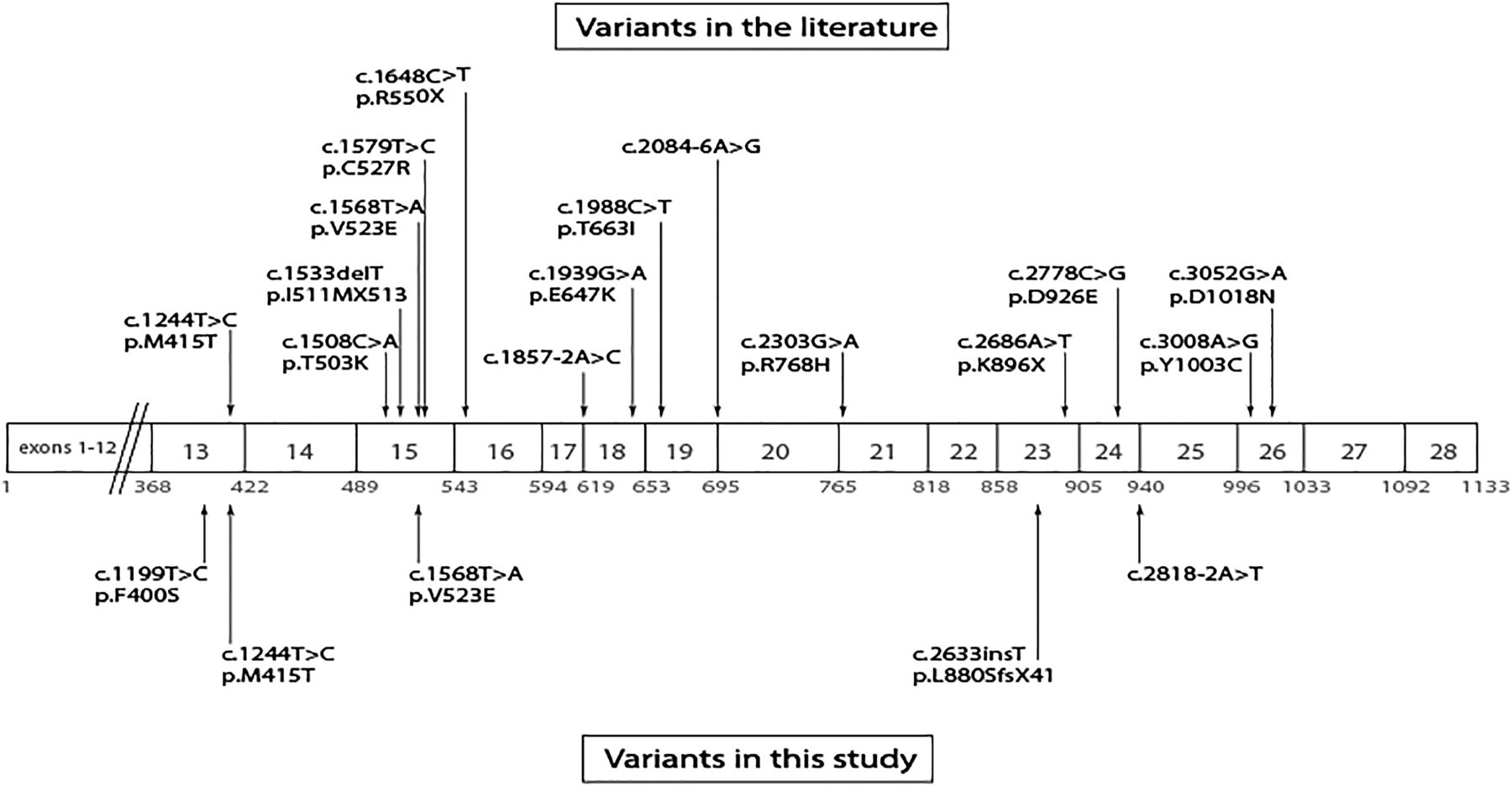
The distribution of these variants across the protein is shown in figure 5. In silico, all of the POLR3B variants were predicted to be deleterious (table 2).
Pathogenicity of the variants identified in the patients determined by various prediction programs
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the POLR3B gene.
In addition to these variants in POLR3B, in subjects #3a and 3b, a rare variant was identified in PROKR2, which has been implicated in hypogonadotropic states (PROKR2 c.[151G>A] p.[A51T]). In silico analysis was not conclusive as two of four of the bioinformatics programs predicted that the variant is tolerated (SIFT, Polyphen2) and the other two predicted it to be deleterious (PROVEAN, Mutation Taster).
Discussion
This report identifies four individuals initially thought to have ‘typical’ hypogonadotropic hypogonadism who were each found to carry two rare variants in POLR3B, a gene implicated in 4H leucodystrophy, which is characterised by ataxia, hypodontia and IHH. The individuals reported in this manuscript have not been found to have variants in any of the other known genes associated with ataxia (POLR3A, POLR1C, RNF216, OTUD4, PNPLA6, STUB1, SQSTM1). Although the vast majority of patients with IHH respond well to exogenous pulsatile GnRH (indicating a hypothalamic defect), subject #1 was resistant to exogenous pulsatile GnRH as demonstrated by failure to raise his pituitary gonadotropins and T level. Although not appreciated at the time, subject #1's pituitary resistance to exogenous GnRH was a clue to his underlying pathophysiology as pituitary defects have been described in other patients with neurodegenerative/reproductive diseases.5 ,8
The identification of four patients with IHH each harbouring two rare variants in POLR3B, and no frank neurological symptoms raises several important concepts—clinically and genetically. Viewed solely within the prism of IHH, these observations demonstrate the complexity of the genetic architecture of this disorder. Over the past several years, numerous genes have been identified for IHH and KS, most of which are involved with GnRH neuronal migration and/or GnRH secretion. Few genes have been identified which cause IHH due to defects singularly affecting the pituitary gland. This small list includes GNRHR, which encodes the GnRH receptor35 ,36 and NROB1, a transcription factor linked to the development of the pituitary gonadotropes and the adrenal gland.37 Therefore, the discovery of mutations in POLR3B in patients with isolated IHH, and no neurological anomalies typical of individuals with two POLR3B variants, suggests that there may be a larger complement of patients whose physiopathology resides at the level of the pituitary, not the hypothalamus. This is an important consideration as most patients with IHH/KS are assumed to have hypothalamic defects; few patients have the opportunity to undergo long-term treatment with exogenous GnRH that is required to reveal a pituitary defect.
In addition to expanding the phenotypical spectrum of 4H leucodystrophy, subject #1's clinical data present some important information about the pace of the reproductive deficiency in this condition. Despite having two variants in POLR3B, neurodegenerative signs still had not appeared by the time of his last evaluation when he was 29 years. At the time of subject #1's initial presentation, he was noted to have robust testicular volume (15 cc) and a subnormal (but clearly measurable) testosterone level, suggesting that his hypothalamic-pituitary-gonadal cascade, while not normal, had supported partial LH and FSH secretion. However, over time, an insidious erosion of gonadotrope integrity must have occurred, as subject #1's clinical investigative studies demonstrated failure to respond to exogenous pulsatile GnRH despite months of dose escalation. This suggests that in patients in whom a genetic risk for this disorder is identified, there may be opportunities for early intervention that could halt or even correct progression of this disease.
Similar to subject #1, subject #2 also presented with IHH; however, she also showed no neurodegenerative signs at the time of her last evaluation when she was in her early 30 s. Subjects #1 and #2 were both found to carry the missense mutation c.[1244T>C] p.[Met415Thr], which has been previously linked to 4H leucodystrophy in multiple individuals carrying c.[1244T>C] p.[Met415Thr] in addition to another missense mutation in POLR3B.38 ,39 The ages of presentation of these patients reported in the literature, along with their symptoms, indicate that they followed a clinical course typical of those with POLR3B mutations. Taken together, these findings raise the possibility that this variant (c.[1244T>C] p.[Met415Thr]) may not be harmful, although the majorityof prediction programs suggest it is pathogenic (PolyPhen2: probably damaging, SIFT: damaging, PROVEAN: deleterious, Mutation Tester: disease causing, PANTHER: not deleterious). Alternatively, it is possible that distinct phenotypes emerge when Met415Thr is ‘partnered’ with different PORL3B transcripts.
Patients #3a and #3b, siblings who presented with absent puberty, were found to have compound heterozygous changes in POLR3B; however, at the time of their last evaluation they had still not shown signs of neurodegeneration. One possibility for their lack of neurodegenerative symptoms may be that the variants identified in POLR3B in these subjects are less harmful to protein function. This appears to be true for c.[1568T>A] p.[(Val523Glu)], which is the most commonly reported rare variant in POLR3B,12 ,13 ,40 and appears to be correlated with later disease onset and a milder clinical course.20 While this variant was predicted to be disease causing or damaging by three out of five programs used for in silico analysis, recent data suggest that these computational tools are more discordant and not as accurate as previously believed.41 Moreover, the PolyPhen2 software tool, which was found to provide the most reliable predictions regarding the impact of amino acid changes to protein structure and function compared with the other programs, predicted that c.[1568T>A] p.[(Val523Glu)] is benign.41 Additional examples supporting the possibility that c.[1568T>A] p.[(Val523Glu)] is not highly damaging do exist in the literature. For example, one of two siblings homozygous for c.[1568T>A] p.[(Val523Glu)] did not show any neurological signs at 21 years of age, in contrast to the majority of patients with biallelic variants in POLR3B who begin to manifest neurological findings in early childhood between the age of 1 year and 2 years.20
In considering why subjects #1–3b with POLR3B variants might have only reproductive and not neurological phenotypes, one issue to consider is the location of the variants within the gene. Previous studies have suggested that exon 15 of POLR3B may be a hot spot for mutations.11 In one study, a third of all variants (including c.[1568T>A] p.[(Val523Glu)], which was identified in this study) were identified as occurring in this specific exon.11 We identified only one variant in exon 15 in the four subjects. RNA polymerase III structure has not been studied to the same extent as RNA polymerase II; however, a number of studies have shown homology between the genes encoding the polymerase subunits POLR3B and POLR2B (87.2%), as well as between POLR3A and POLR2A (83.2%).9 ,15 ,42–46 Extrapolating from the reported domains of POLR2B, the POLR3B variants that were identified in this study are distributed in three different functional domains; in other words, they are not clustered in a single domain that might provide an explanation for the restricted reproductive phenotype of subjects #1–3.
In a recently published article, a subset of patients with 4H leucodystrophy and mutations in POLR3A or POLR3B were lacking the diffuse hypomyelination generally associated with mutations in these genes.39 Despite the absence of hypomyelination in these patients, they were all observed to have ataxia, a symptom understood to be characteristic of these mutations and 4H leucodystrophy. In contrast, the four individuals reported in this manuscript were not found to have any signs of neurodegeneration or ataxia by the time of their last evaluations in their early 30 s, despite their two variants in POLR3B. Their diagnoses of hypogonadotropic hypogonadism without the other symptoms of 4H leucodytrophy broadens the understanding of this spectrum of disorders even further.
One of the limitations of this study is length of follow-up. All four individuals presented much later in life than the typical patient with 4H leucodystrophy who presents in infancy/early childhood. It is possible that these subjects may escape neurological sequelae for years to come; however, reaching that conclusion will require careful longitudinal follow-up. As summarised above, neurological abnormalities were not apparent during clinical and research evaluations (by multiple providers), but it is always possible that subtle signs were missed.
Thus, the spectrum of phenotypes resulting from POLR3B mutations is wider and more varied than previously thought. Patients carrying mutations in POLR3B may present with reproductive failure due to hypogonadotropism and no neurological or dental defects. Further analyses of additional genes implicated in ataxia/hypogonadal syndromes will likely further illuminate the complexity of these disorders. As the concept of personalised medicine grows and becomes increasingly adapted into our clinic practices, understanding the full phenotypical spectrum of genotypes associated with these complex syndromes will be increasingly important.
Acknowledgments
The authors thank the research subjects and members of the Massachusetts General Hospital Reproductive Endocrine Unit for discussions and reading of the manuscript. The authors also thank Dianali Rivera Morales for her help in editing the manuscript and Jenny Jing for her help in sequencing samples.
References
Footnotes
Contributors SBS was responsible for the concept and design of the study. MRR and SBS drafted the main manuscript. LP and SBS analysed and interpreted the data. RQ and PK contributed clinical data. Y-MC, MFL and LP revised the manuscript and made comments on the structure, details and grammar of the article.
Funding The research was supported by the National Institutes of Health Eunice Kennecy Shriver National Institute of Child Health and Human Development (P50HD028138).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Partners Human Research Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional phenotype or genotype information may be requested by contacting SBS.