Article Text

Abstract
Background Agalsidase β is a form of enzyme replacement therapy for Fabry disease, a genetic disorder characterised by low α-galactosidase A activity, accumulation of glycosphingolipids and life-threatening cardiovascular, renal and cerebrovascular events. In clinical trials, agalsidase β cleared glycolipid deposits from endothelial cells within 6 months; clearance from other cell types required sustained treatment. We hypothesised that there might be a ‘lag time’ to clinical benefit after initiating agalsidase β treatment, and analysed the incidence of severe clinical events over time in patients receiving agalsidase β.
Methods The incidence of severe clinical events (renal failure, cardiac events, stroke, death) was studied in 1044 adult patients (641 men, 403 women) enrolled in the Fabry Registry who received agalsidase β (average dose 1 mg/kg every 2 weeks) for up to 5 years.
Results The incidence of all severe clinical events was 111 per 1000 person-years (95% CI 84 to 145) during the first 6 months. After 6 months, the incidence decreased and remained stable within the range of 40–58 events per 1000 patient-years. The largest decrease in incidence rates was among male patients and those aged ≥40 years when agalsidase β was initiated.
Conclusions Contrary to the expected increased incidence of severe clinical events with time, adult patients with Fabry disease had decreased incidence of severe clinical events after 6 months treatment with agalsidase β 1 mg/kg every 2 weeks.
Trial registration number NCT00196742.
- Cardiovascular Medicine
- Clinical genetics
- Getting Research into Practice
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Fabry disease (OMIM #301500) is a rare, X linked, multisystemic, multiorgan disorder in which globotriaosylceramide (GL-3) and other glycosphingolipids accumulate within lysosomes due to deficient activity of α-galactosidase A (α-GalA).1 ,2 The progressive accumulation of GL-3 in multiple cell types is ultimately associated with severe, potentially life-threatening target-organ complications,3 including cardiac events,4 stroke,5 renal failure6 and premature death.7 ,8
The spectrum of clinical presentations is wide and includes the severe classic male phenotype, later-onset variant phenotypes and variable clinical presentations in female patients ranging from asymptomatic to, occasionally, a severe classic phenotype.1 ,2 ,9
Since 2001, enzyme replacement therapy (ERT) has been available for treating patients with Fabry disease.10 ,11 Agalsidase β (Fabrazyme; Genzyme, a Sanofi company, Cambridge, Massachusetts, USA), a form of recombinant human α-GalA, is approved for use as ERT in Fabry disease in the USA, Europe and many other countries throughout the world. Clinical trials of agalsidase β at dose 1 mg/kg every 2 weeks have shown that treatment results in the complete clearance of GL-3 deposits in the vascular endothelial cells of the kidneys, heart and skin at 6 months in the majority of adult patients, thereby reducing the overall glycolipid burden,10 ,12 although minimal clearance was observed in cardiomyocytes and podocytes.12 ,13
Due to the relatively recent availability of ERT, and the commonly occurring delays in diagnosis,14 ,15 ERT has been initiated in many patients after substantial target organ damage has occurred. Not unexpectedly, therefore, serious Fabry disease-related clinical events have occurred in these patients despite ERT16–18 and the use of ERT for patients with advanced kidney disease has been questioned.19 The key question, however, is not if ERT completely prevents severe clinical events in patients with Fabry disease, but whether ERT reduces the incidence and/or progression of severe clinical events. Given the difficulty and ethical concerns of conducting long-term placebo-controlled clinical trials in patients with Fabry disease, patient registry data are needed to gain insights into the effect of ERT on the incidence rates of severe clinical events.
In this study, we analysed data from adult patients with Fabry disease enrolled in the Fabry Registry (ClinicalTrials.gov identifier: NCT00196742) with the objective of determining trends in the incidence of severe, potentially life-threatening clinical events over time in patients receiving up to 5 years treatment with agalsidase β, at a dose of 1 mg/kg every 2 weeks. The analysis was limited to adult patients, as severe clinical events are relatively uncommon in childhood due to the relatively slow disease progression observed in the majority of patients with Fabry disease.
Methods
Study design
The Fabry Registry was initiated in 2001 as part of an initiative to help healthcare professionals involved in the treatment or diagnosis of Fabry disease better understand the disease and its management, and to help create treatment monitoring guidelines for the disease. Patient participation in the Fabry Registry is voluntary and the total number of patients with Fabry disease who will participate in the Fabry Registry and the associated person-years of follow-up are not predefined. It is at the discretion of the treating physician to determine the testing protocol for each patient.
Inclusion/exclusion criteria
To be included in these analyses, Fabry Registry adult patients must have received agalsidase β as their initial source of ERT given at or near the recommended licensed dose of 1 mg/kg every 2 weeks (average dose range: ≥0.9 to <1.1 mg/kg every 2 weeks) for up to 5 years. Adult patients aged ≥18 years at ERT initiation were eligible for this analysis. Patients who developed end stage renal failure (signified by dialysis or transplant) before starting agalsidase β were excluded because they have already had an irreversible organ failure. Fabry Registry patients with other pretreatment clinical events were included in our analysis. Patients with non-classic, later-onset variant mutations (p.Asn215Ser (N215S), p.Arg112His (R112H), p.Ala97Val (A97V), p.Gly35Arg (G35R), p.Ile317Thr (I317T), p.Arg301Gln (R301Q); n=56),20–25 as reported in the ‘fabry-database.org’ Fabry mutation database,26 ,27 were excluded from the analysis. The ‘fabry-database.org’ database of clinical phenotypes, genotypes and structures of mutant GLAs has been built by the research group led by Dr H Sakuraba in order to help researchers and clinicians who study Fabry disease. The database currently contains more than 640 different mutations.27 Moreover, individuals with non-pathogenic Fabry gene polymorphisms (p.Asp313Tyr (D313Y), p.Arg118Cys (R118C); n=3)28–30 were excluded. No patients in the cohort had a p.Ala143Thr or IVS4+919G>A mutation, or the polymorphism p.Glu66Gln (E66Q). The analysis population included: (1) patients with mutations categorised in the ‘fabry-database.org’ Fabry mutation database as being associated with ‘classic’ Fabry disease; (2) patients with mutations not entered or classified in this database; (3) patients for whom mutations were not reported in the Fabry Registry.
Outcomes and follow-up
Death due to any cause, and the following three severe (potentially life-threatening) clinical event categories were included in the analysis: (A) renal events: chronic dialysis (>40 days) or renal transplantation; (B) cardiovascular events: myocardial infarction, first-time congestive heart failure, atrial fibrillation, ventricular tachycardia, evidence of progressive heart disease sufficiently severe to require a pacemaker, heart bypass surgery, coronary artery dilatation or implantation of a cardioverter/defibrillator; and (C) cerebrovascular events: haemorrhagic or ischaemic stroke. The outcome was defined as the time to death or the first event in any of the above predefined categories. Exposure time started when each patient began agalsidase β treatment, and ended at the time of their first reported severe clinical event after starting ERT, when they stopped receiving agalsidase β (due to treatment interruption or switch to another source of ERT), or at the last follow-up visit. Follow-up data were not included beyond 25 June 2009 because many patients began temporary agalsidase β dose-reductions due to an interruption in the manufacturing process and a subsequent global shortage of agalsidase β around this time. Follow-up time was censored at 5 years on agalsidase β treatment because in 2009 relatively few patients had been on ERT for longer than 5 years.
Statistical methods
Descriptive statistics were used to summarise the demographic and medical characteristics of patients at the time of agalsidase β initiation. Continuous variables were compared using the Wilcoxon test and categorical variables were compared using the χ2 or Fisher's exact test, when appropriate. For the incidence rate tables, the rate for the first event per 1000 patient-years of follow-up time and the associated 95% CIs were computed for each time period on treatment.31 Total patient-years of follow-up time was calculated as the sum of follow-up times for each patient within each specified category. Incidence rates were calculated for all patients and further stratified by: (A) non-renal event prior to first agalsidase β infusion (‘pre-ERT event’); (B) median age at initiation of agalsidase β treatment; and (C) gender. Finally, Cox proportional hazards regression models were used to evaluate the impact of risk factors associated with severe clinical events, including a severe non-renal event prior to first ERT, age at agalsidase β initiation and gender. The small number of excluded individuals with Fabry gene polymorphisms or later-onset variant mutations did not allow for a meaningful separate analysis of these groups to assess time to treatment benefit in these groups. Statistical analyses were performed using SAS statistical software V.9.2 (SAS Institute, Cary, North Carolina, USA). An α level of 0.05 was used as the criterion for statistical significance.
Results
Patient disposition
As of 25 June 2009, 1044 adults with Fabry disease (641 men, 403 women) treated for up to 5 years with agalsidase β (average dose range: ≥0.9 to <1.1 mg/kg every 2 weeks) were included in the present analysis. Mean (SD) time on agalsidase β was 2.8 (1.8) years with a maximum follow-up of 5 years. The median age at which agalsidase β was initiated was 40 years (IQR: 31–50 years) and it was used for further subgroup analysis.
Baseline patient demographics
The baseline characteristics at the time of initiation of agalsidase β treatment are shown in table 1. Many patients had advanced disease with a frequency of pre-ERT events of 17% , left ventricular hypertrophy (LVH) 61%, and an estimated glomerular filtration rate (eGFR) <60 mL/min/1.73 m2 of 24%. Furthermore, patients who started agalsidase β treatment at age ≥40 years had more evidence of renal and cardiac organ damage compared with patients who started treatment at age <40 years (table 1). In patients who started ERT at age ≥40 years, the proportion of patients with eGFR <60 mL/min/1.73 m2 was 33% compared with 15% of those who initiated ERT at age <40 years. Similarly, more patients who started treatment at age ≥40 years had a baseline urine protein:creatine ratio of >0.5 g/g (54% vs 38%), LVH (79% vs 37%), arrhythmias (30% vs 17%), systolic blood pressure >130 mm Hg (47% vs 35%) and diastolic blood pressure >80 mm Hg (46% vs 30%) compared with those who started ERT at age <40 years. Missense (42%) and nonsense mutations (14%) were most commonly reported, followed by frameshift mutations (10%). Genotype data were not available for 27% of the patients (see online supplementary table S1). Four hundred patients had mutations categorised in the ‘fabry-database.org’ database as being associated with classic Fabry disease, whereas 365 patients had mutations not entered or classified in this database.27
Baseline characteristics of adult patients with Fabry disease receiving agalsidase β (1 mg/kg every 2 weeks) as their initial source of ERT and included in the study
Supplementary tables
Type and timing of first severe clinical events following initiation of ERT
Overall, 177 of 1044 (17%) patients had a severe clinical event within 5 years of agalsidase β treatment initiation: 120 events occurred in 641 male patients (19%) and 57 events occurred in 403 women (14%). For all patients, cardiac and renal events were the most common, followed by strokes and death. The absolute number of events, defined by clinical category over time following initiation of ERT for men and women are shown in online supplementary figure S1. In 54 of 177 (31%) patients with severe clinical events, the first event occurred during the first 6 months of ERT. The pattern of first event occurrence appeared similar across individual event types, except for death which was uncommon throughout the maximum follow-up interval. The event subtypes together with the overall frequencies of occurrence are shown in the online supplementary table S2. Of the cardiovascular events, ventricular tachycardia (2%) contributed the least to the first event in the composite (other cardiovascular events: myocardial infarction 7%, first-time congestive heart failure 6%, atrial fibrillation 8%, evidence of progressive heart disease sufficiently severe to require a cardiac procedure 16%).
Supplementary figure
Absolute number of patients experiencing first clinical event by time on agalsidase beta: male patients (A); and female patients (B). Note: the two columns near the vertical axis correspond to shorter time periods (6 months) than the other four columns (12 months).
Incidence of combined severe clinical events over time following initiation of ERT
Clinical events were combined when calculating incidence rates, as there were too few events to analyse incidence rates for individual categories (cardiac, stroke, renal and death). The incidence rate was 111 events per 1000 patient-years (95% CI 84 to 145) during the first 6 months, followed by decreased incidence rates with longer exposure to agalsidase β (figure 1). After 6 months of ERT, the incidence rate for severe clinical events decreased and remained stable within the range of 40–58 events per 1000 patient-years for the rest of the follow-up interval.

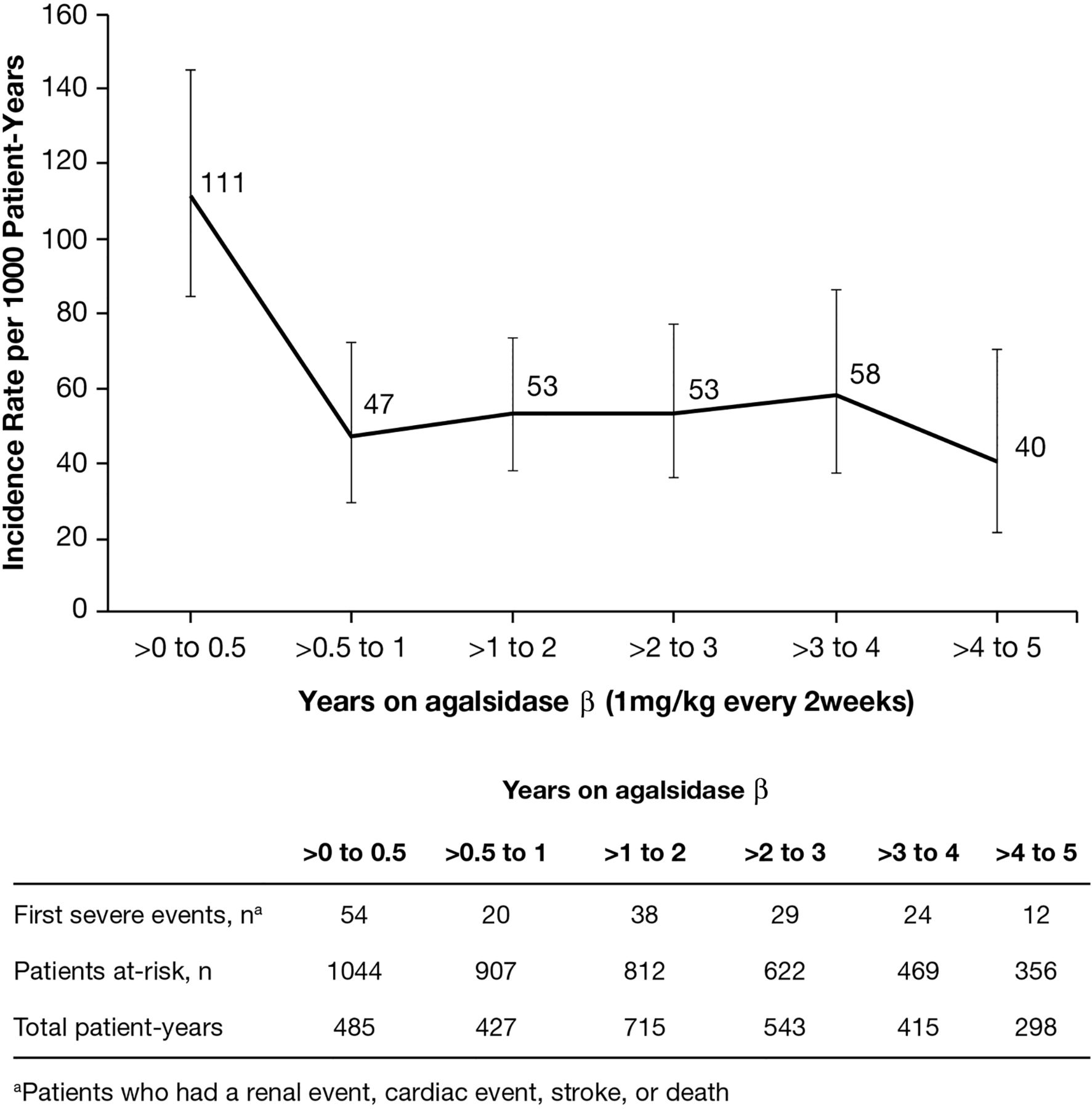{kind=link}
Incidence rate of severe clinical events (rate per 1000 patient-years with 95% CIs) over time following initiation of enzyme replacement therapy.
Incidence of combined clinical events by history of pre-ERT event, age and gender
Given that Fabry disease is clinically diverse, incidence rates of clinical events were stratified by history of pre-ERT clinical event, median age at agalsidase β initiation and gender (table 2). The number of years on ERT was presented in three categories: 0–0.5 year, >0.5–1 year and >1–5 years. There was attenuation of the incidence rates of severe clinical events after the first 6 months of ERT. Differences between strata were more pronounced among patients with high risk of events (ie, age ≥40 years when ERT was started or male sex) during the first 6 months of ERT (table 2).
Incidence rates of severe clinical events (renal event, cardiac event, stroke or death) per 1000 patient years while on agalsidase β by history of pre-ERT event, age and sex (177 severe clinical events were observed during 5 years maximal follow-up)
Among patients with a pre-ERT event, the incidence rate during the first 6 months was 166 clinical events per 1000 patient-years (95% CI 88 to 283). The clinical event rate decreased after 6 months to 102 per 1000 patient-years (95% CI 41 to 210) during the 0.5–1 year period and remained at 102 per 1000 patient-years (95% CI 68 to 146) during the >1–5 years period. Among patients aged ≥40 years at ERT initiation, the incidence of clinical events during the first 6 months was 177 events per 1000 patient-years (95% CI 128 to 239), which decreased to 74 per 1000 patient-years (95% CI 42 to 123) during the >0.5–1 year period and 79 per 1000 patient-years (95% CI 61 to 99) during the >1–5 years period. Among men, the incidence rate during the first 6 months was 124 per 1000 patient-years (95% CI 87 to 171), which decreased after this time period to 49 per 1000 patient-years (95% CI 26 to 85) during the >0.5–1 year period and 54 per 1000 patient-years (95% CI 42 to 68) during the >1–5 years period. There were too few clinical events to meaningfully analyse the patient subgroups based on disease severity.
Risk factors for severe clinical events
The results of the Cox proportional hazards regression analysis assessing the association of risk factors with clinical events are shown in table 3. Among patients with pre-ERT clinical events, the risk of developing an event after starting agalsidase β was not significantly different during 0–0.5 year compared with those who had no previous clinical events (Model 1: HR 1.1, 95% CI 0.6 to 2.0, p=0.81), but increased thereafter (Model 2: HR 1.8, 95% CI 1.2 to 2.7, p<0.01) (table 3). Compared with patients aged <40 years at first ERT, patients aged ≥40 years at first ERT had a significantly higher risk of having an event during 0–0.5 year (Model 1: HR 4.4, 95% CI 2.2 to 8.7, p<0.01), but this decreased during the period >0.5–5 years (Model 2: HR 2.5, 95% CI 1.7 to 3.8, p<0.01). Compared with female patients, the risk of a clinical event in male patients was significantly higher during 0–0.5 year (Model 1: HR 1.9, 95% CI 1.1 to 3.4, p=0.03), but not thereafter (Model 2: HR 1.5, 95% CI 1.0 to 2.1, p=0.06). There was no evidence of a violation in the proportional hazards assumption with the number of events available for this analysis.
Cox proportional hazards regression analysis assessing the time dependence of risk factors for clinical events*
Discussion
Severe clinical events are hard end points that may provide stronger evidence of clinical benefit with agalsidase β than surrogate end points such as clearance of GL-3 deposits, degree of LVH or changes in renal function (eg, eGFR). Our analysis of Fabry Registry data provides new information on the incidence of severe clinical events over time for adult patients treated with agalsidase β. We show that the incidence rate for severe clinical events decreases after the first 6 months of treatment with agalsidase β. Those patients at highest risk for severe clinical events, due to older age or male sex, displayed the greatest absolute reduction in event rate over time on agalsidase β, and in particular had the greatest reduction in the incidence rates for severe clinical events after the first 6 months of treatment. These results are notable because the occurrence of severe clinical events, despite ERT, has recently been emphasised16 ,17 and the efficacy of ERT for patients with more Fabry-related kidney involvement has been questioned.19
Our observation of a decrease in severe events over time is consistent with findings from a phase IV trial in which 82 adults with advanced Fabry disease were randomised to either agalsidase β 1 mg/kg every 2 weeks (mean age 47 years) or placebo (mean age 44 years).32 After a median follow-up period of 18 months, the incidence of a first severe clinical event was reduced for patients treated with agalsidase β compared with those in the placebo group (HR 0.47, 95% CI 0.21 to 1.03).32 Together these data support the hypothesis that treatment with agalsidase β reduces the rate of severe clinical events, rather than the alternative potential explanation that ERT causes an absolute increase in the short-term incidence of severe clinical events. In this regard, a single-centre study of 57 ERT-treated patients observed that, in comparison to a historical group of untreated patients, the odds for development of a first severe clinical event declined with longer treatment duration per year of ERT (OR 0.81, 95% CI 0.68 to 0.96).17
In the general population, ageing is a known risk factor for the development of cardiac, renal and stroke events and a prior cardiac or stroke event is associated with a higher risk of a second cardiac or stroke event.33 ,34 In patients with Fabry disease, the natural course of the disease is also associated with an increasing number of these events as the severity of organ involvement increases as patients age with cardiac disease being the leading cause of death.7 ,8 Thus, the observed decrease in the incidence rate of severe clinical events during the first 5 years of agalsidase β treatment is important and consistent with a modifying effect of ERT on the course of Fabry disease in adults.
In the present cohort, the occurrence of a severe clinical event prior to the initiation of agalsidase β was associated with a higher risk of experiencing another serious event while receiving treatment after the first 6 months of treatment. This finding was observed even though we excluded patients who had a severe renal event leading to organ failure (dialysis or transplantation) prior to starting ERT. Recent studies have emphasised the associated risks for mortality, stroke and coronary heart disease associated with albuminuria and eGFR <60 mL/min/1.73 m2 in the general population.35–37 These same risk factors are also evident in the baseline characteristics of patients with Fabry disease described in this report. However, patients who had a severe clinical event prior to ERT initiation may have subsequently been given appropriate adjunctive therapy, which may result in a lower estimation of the subsequent event rates in this group.
In the current analysis, many patients started agalsidase β relatively late in the course of their disease, which reflects the delay in diagnosis of Fabry disease and the availability of ERT.14 ,15 Indeed, the baseline characteristics of the patients included in our study demonstrate a higher prevalence of advanced disease in those aged ≥40 years before ERT initiation. Data from a previous Fabry Registry study showed that 81% of 52 patients with classic Fabry disease, who originally participated in a randomised phase III clinical trial, did not experience any severe clinical events during a median 10-year period of agalsidase β treatment.38 Several studies have also shown that patients who initiate treatment at a younger age, and with less kidney involvement, benefit the most from therapy in terms of changes in white matter lesions, left ventricular mass and surrogate outcomes such as decline in eGFR.38–41
Delayed time between an intervention and the time at which improved health outcomes/clinical benefits are seen leads to ‘lag time’ bias, and if this effect is not considered when results are analysed, there may be inappropriate pessimism with cost-effectiveness analyses of the benefits of new therapies.42 The results from our analysis are consistent with a delay or lag time of 6 months from initiation to clinical benefit from agalsidase β. There are several recent examples of delayed time to benefit varying from 6 months for statin therapy in secondary prevention of cardiovascular effects, to >10 years in prostate cancer screening or intensive insulin therapy.43–46 For example, the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study in diabetic kidney disease recently published results with a median follow-up of 22 years.44 It was shown that intensive insulin therapy was associated with a lower long-term risk of an impaired eGFR, but that the curves only began to differ after a decade of follow-up.44
For Fabry disease, we can only speculate as to the pathophysiological effects underlying the observed decreased incidence of severe events during treatment with agalsidase β. A placebo-controlled randomised clinical trial reported that 70% of patients treated with 1 mg/kg agalsidase β have nearly complete clearance of kidney microvascular endothelial glycolipid deposits after 6 months.10 ,12 Thus, the lag time to benefit observed in our analysis is consistent with the time course of GL-3 clearance from vascular endothelial cells. Although GL-3 clearance of additional cell types, such as podocytes and cardiomyocytes, takes longer than 6 months,12 ,47 ,48 clearance of vascular endothelial cells may be significant in terms of endothelial cell biology and as a marker of overall glycolipid burden. Thus, we speculate that the lag time to benefit depends on significant glycolipid clearance from endothelial cells and the associated subsequent reversal of endothelial cell dysfunction.
The patient population in our study is representative of adult patients receiving ERT with respect to age, gender18 ,41 and genotype (see online supplementary tables S1 and S3). Considering that the patient population is representative of patients with Fabry disease in the clinic, the findings of the current analysis may inform therapeutic decision-making in these patients. While our findings emphasise the importance of starting ERT at a relatively young age, we also clearly demonstrate a benefit of ERT in older patients with Fabry disease with more advanced disease who better represent the majority of patients reported to date.19 ,49
Fabry disease is a clinically heterogeneous disease and little is known about the natural progression of Fabry disease in patients with later-onset variant mutations and reference studies in which ERT-treated patients with these mutations and classically affected patients have been analysed separately are lacking. Therefore, the main analysis population was restricted to patients with mutations categorised as being associated with classic Fabry disease, patients with mutations not entered or classified in the ‘fabry-database.org’ database,27 and patients for whom mutations were not reported in the Fabry Registry (27% of the patients); even though these mutations were not recorded in the Fabry registry, the treating physicians classified these patients as ‘classical’ Fabry disease. The frequencies of severe events in the latter two groups of patients (117 events in 644 patients (18%)) and in patients with mutations associated with classic Fabry disease (60 events in 400 patients (15%)) were comparable (see online supplementary table S3). Most patients have a positive family history (data not shown), and all received ERT suggesting that disease severity criteria for treatment initiation, as per published general guidelines or local/national protocols, had been met. Taken together, the results support the inclusion of patients with unclassified or missing mutations. While the percentage of patients with available mutation data included in the current analysis is relatively high (73%), a priority of the Registry is to increase the amount of mutation data entered.
Several other limitations and cautions need to be considered when interpreting the results. The Fabry Registry is not a clinical trial and interpretation of these findings is limited by the lack of an appropriate parallel-untreated control group due to the absence of a well-matched contemporaneous group of patients. Patient participation in the Fabry Registry is voluntary and not all patients with Fabry disease have been identified; also, not all patients will wish to participate. A limitation of any registry includes a possible patient selection bias toward the inclusion of more severely affected treatment-naïve or treated patients. In addition, event surveillance may be more comprehensive immediately following institution of ERT, which could explain in part the results reported herein. Substantially more men than women are included in this study; thus, the results for female patients apply primarily to women with manifestations of Fabry disease that were severe enough to have been treated with ERT. Furthermore, although the Fabry Registry provides a recommended schedule of assessments, patients and their treating physicians ultimately determine the timing of assessments and the time intervals at which they are carried out. Local standards of care may differ between treatment sites and across all time points, especially with respect to the use of vasculoprotective and renoprotective adjunctive therapies. Thus, clinical data may be incomplete for some patients and some assessments or events may be under-reported; additionally, some patients may be lost to follow-up. The absolute event rates are low overall, so that caution is warranted in interpreting the event rate in any individual short interval, as the CIs for the incidence rates are increased for short follow-up intervals.
Serum-mediated inhibition of ERTs is emerging as an important issue particularly in male patients with classic Fabry disease.50 No systematic evaluation of this issue was available for inclusion in the manuscript. Incompleteness of the data set for the temporary dose reduction period precluded a meaningful analysis of clinical events occurring after dose reductions. The focus of the current analyses was: (A) first severe clinical event in order to understand primary prevention associated with agalsidase β treatment; (B) a time frame of up to 5 years on agalsidase β, as the number of patients treated for longer than 5 years was very low; and (C) patients treated with the approved dose and formulation of agalsidase β. Further research is needed to understand outcomes for longer periods of time or other doses/formulations of ERT. A major strength of this analysis is that it represents the largest longitudinal follow-up data set and evaluation of time-to-event analysis for severe clinical events in adult patients with Fabry disease treated with agalsidase β.
In conclusion, the occurrence of severe clinical events in patients with Fabry disease decreases after the first 6 months of initiation of treatment with agalsidase β. This is in contrast to the expected increase in events due to ageing. Interestingly, the decrease in the incidence of severe clinical events with time on agalsidase β was most conspicuous for those populations at greater risk of events because of older age or male gender. These observations are also consistent with a delayed benefit after initiation of agalsidase β in patients with Fabry disease, which is similar to the time needed to clear vascular endothelial cells of GL-3 deposits with ERT. Although the small number of events precluded further subgroup analysis, older age (≥40 years) at initiation of treatment was associated with more advanced disease. The apparent clinical benefit of ERT for serious clinical events was observed even in patients with more advanced disease, supporting prior clinical trial results32 and providing an alternative perspective to recent recommendations for withholding ERT in patients with more advanced disease.19 Time-to-event analysis will be important in future studies that compare different doses or preparations of ERT, or other modalities for treating Fabry disease.49
Acknowledgments
The authors thank the patients who agreed to participate in the Fabry Registry, as well as the physicians and research coordinators who have entered clinical data on behalf of these patients. The authors thank colleagues at Genzyme, a Sanofi company, for medical input (Wytske Kingma), epidemiological advice (Judy Kempf), programming support (Badari Gudivada) and copy editing (Cheryl Lathrop). The authors received editorial/writing support in the preparation of this manuscript provided by Niina Nuottamo of Excerpta Medica, funded by Genzyme, and Hans Ebels of Genzyme, a Sanofi company. The authors were responsible for all content and editorial decisions, and received no honoraria related to the preparation of this publication.
References
Footnotes
Contributors Analysis and interpretation of data: AO, SSM and DGW. Drafting the article: AO and DGW. All authors contributed to acquisition of data, reviewing and revising the article critically for important intellectual content, final approval of the version to be published, agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This research was funded by Genzyme, a Sanofi company. AO was supported by ISCIII REDINREN RD012/0021 and Intensification FEDER funds.
Competing interests AO is a consultant for Genzyme and has received speaker fees from Shire HGT. AA is a member of the North American Fabry Registry Board and has received speaker fees for lectures on Fabry disease from Genzyme. DGB is a member of the North American Fabry Registry Board and has received speaker fees for lectures on Fabry disease and for clinical studies from Genzyme, Shire HGT and Amicus Therapeutics. GC has active research support and consulting arrangements with Genzyme. JC has received honoraria for consultation and advisory board participation from Genzyme, Shire HGT, Pfizer Corporation/Protalix Corporation and Synageva BioPharma. He has been an investigator in clinical trials sponsored by Genzyme, Shire HGT, Biomarin Pharmaceuticals and Amicus Therapeutics. He has received honoraria for lectures from Genzyme, the National Gaucher Foundation, the Fabry Support and Information Group, and the SIMD North American Metabolic Academy. DPG has been in receipt of honoraria for lectures on Fabry disease from Genzyme, Shire HGT and Amicus Therapeutics. RJH consults with Genzyme and Shire HGT, and has been an investigator in clinical trials sponsored by Genzyme, Shire HGT and Amicus Therapeutics. These activities have been monitored and found to be in compliance with the conflict of interest policies at Cincinnati Children's Hospital Medical Center. AJ is a member of the Fabry Registry Board. She has received advisory board fees and honoraria for lectures on Fabry disease from Genzyme, Shire HGT and Amicus Therapeutics. AL is a member of the Fabry Registry Board. He has been in receipt of honoraria for lectures on Fabry disease from Genzyme Corp, Shire HGT and Amicus Therapeutics. SSM is an employee of Genzyme. MM consults for Genzyme. This interest has been reviewed and managed by the University of Minnesota in accordance with its conflict of interest policy. He also consults for Amicus Therapeutics and has reviewed grants for Shire HGT. JPO has received travel support from Genzyme, Shire HGT and Amicus Therapeutics, and speakers fees and research support from Genzyme. MRP has received advisory board fees from Genzyme, Jansen, Bayer and Merck; and research grants from NHLBI, AHRQ, AstraZeneca, CSI and Maquet. JP has been in receipt of honoraria for lectures on Fabry disease from Genzyme, Shire HGT, Amicus Therapeutics and Protalix Corporation. SW is a member of the Fabry Registry Board. He has received honoraria from Genzyme, Shire HGT and Amicus Therapeutics; and fees for consulting from Genzyme and Shire HGT. CW is a member of the European Fabry Registry Board. He has received honoraria from Genzyme and a research grant to the institution. H-WY is a member of the Fabry Registry Board. He has received honoraria from Genzyme. DGW is a consultant for Genzyme, has served as a consultant for Shire HGT and Amicus Therapeutics, and has received research funding from Genzyme and Amicus Therapeutics. These activities have been reviewed and managed by the University of Alabama at Birmingham in accordance with its conflict of interest policy.
Ethics approval Patient participation in the Fabry Registry (NCT00196742) is voluntary. Each independent global site is responsible for obtaining patient's informed written consent to submit his/her health information to the Registry, and to use and disclose this information in subsequent aggregate analyses. The Registry protocol, informed consent form, any locally required authorisation documents to send patient information to the Registry, and relevant supporting documents are reviewed and approved by the local fully constituted Institutional Review Board (IRB) or Independent Ethics Committee (IEC), unless the site provides documentation to the Registry that such review is not required under local country laws or has been waived by a particular IRB/IEC.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Fabry Registry Boards of Advisors are responsible for the scientific oversight of the Fabry Registry. Patient and physician confidentiality is protected within the Fabry Registry. Any healthcare professional enrolled in the Fabry Registry may submit a data analysis request for aggregated analysed data from the Fabry Registry. All requests are reviewed by the Fabry Data Request Review and Decision Committee. Determination of whether a request can be completed or not depends on several factors, including data availability, and analysis scope and feasibility. Data request forms can be found in the RegistryNXT! Library at https://www.registrynxt.com. All relevant data are reported within the paper.