Article Text

Abstract
Background Adolescent idiopathic scoliosis exhibits high heritability and is one of the most common spinal deformities found in adolescent populations. However, little is known about the disease-causing genes in families with adolescent idiopathic scoliosis exhibiting Mendelian inheritance.
Objective The aim of this study was to identify the causative gene in a family with adolescent idiopathic scoliosis.
Methods Whole-exome sequencing was performed on this family to identify the candidate gene. Sanger sequencing was conducted to validate the candidate mutations and familial segregation. Real-time QPCR was used to measure the expression level of the possible causative gene.
Results We identified the mutation c.2645A>C (p.E882A) within the AKAP2 gene, which cosegregated with the adolescent idiopathic scoliosis phenotypes. AKAP2 is located in a previously reported linkage locus (IS4) on chromosome 9q31.2–q34.2 and has been implicated in skeletal development. The mutation was absent in dbSNP144, ESP6500 and 503 ethnicity-matched controls. Real-time QPCR revealed that the mRNA expression level in the patients was increased significantly compared with the family controls (p<0.0001).
Conclusions AKAP2 was therefore implicated as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. Further studies should be conducted to validate the results from the perspective of both the genetics and pathogenesis of this disease.
- adolescent idiopathic scoliosis
- whole-exome sequencing
- AKAP2
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Adolescent idiopathic scoliosis (AIS) is a common, complex, three-dimensional spinal deformity that may occur from the age of 10 to skeletal maturity.1 The prevalence of AIS is 2%–4% in adolescents, with an obvious sex bias.2 The prevalence in females is higher than in males, especially in more severe cases.3
In recent years, several aetiological factors have been associated with AIS, such as hormonal disturbances,4 developmental neuromuscular dysfunction (paraspinal muscle) and so on.5 Nevertheless, no factor has been convincingly shown to be an actual aetiological factor for AIS. Several studies have noted familial clustering of AIS, autosomal-dominant and X-linked of inheritance have been observed.6 ,7 Therefore, AIS is widely accepted to be a disease for which hereditary or genetic factors likely play key roles.
Many genetic studies regarding AIS have previously been reported. Family-based linkage analyses have revealed several AIS linkage loci, such as 19p13.3 (IS1), 17p11.2 (IS2), 8q12 (IS3), 9q31–q34 (IS4) and 17q25-qter (IS5),8–11 however, only the POC5 gene has been identified as causing idiopathic scoliosis.12 Moreover, SNPs in GPR126, CHL1 and LBX1 genes and so on have been documented to be associated with AIS risk.13 ,14 However, only polymorphisms near the LBX1 gene have successively been replicated in different ethnic groups. All these findings suggest that AIS is a disease with high genetic heterogeneity.
In this study, we identified a mutation within the AKAP2 gene that cosegregated with the phenotypes of a Chinese AIS family with an autosomal-dominant inheritance using whole-exome sequencing.
Materials and methods
Subjects and clinical examinations
An AIS family from the Chinese Han population participated in our study. Ten subjects (five affected) were recruited and underwent detailed clinical examinations. All members of the family were ascertained and examined by two experienced orthopaedic surgeons. Diagnostic criteria of AIS were determined through history and physical examinations, including a complete neurological examination and standing anteroposterior/lateral X-ray films of the entire spine. AIS is defined as a lateral curvature of the spine greater than 10° with vertebral rotation and no congenital deformity. Peripheral blood from all members were collected after signing informed consent. In addition, 503 ethnicity-matched subjects were enrolled as controls. This study was approved by the institutional review board of the State Key Laboratory of Medical Genetics.
Whole-exome sequencing and analysis
Peripheral blood lymphocytes were collected for all of the recruited family members. Genomic DNA was extracted using the standard phenol–chloroform method. Three individuals, including two affected cases (IV10, IV11) and one family control (III6), were selected for whole-exome sequencing. Approximately 1 µg of DNA from each sample was used to construct an exome library using the SureSelectXT Target Enrichment System for Illumina Paired-End Sequencing Library kit from Agilent Technologies (Santa Clara, California, USA), following the manufacturer's instructions (V.1.3.1). An Illumina Solexa GAIIx sequencer was used to sequence the prepared library using a paired-end 76 bp×2 method according to the manufacturer's instructions.
The generated bcl files were converted to fastq files using the configureBclToFastq function of the Illumina CASAVA programme (Illumina, San Diego, California, USA), and this step included a basic evaluation of the sequencing quality. We then used the FastQC (V.0.10.0) tool to evaluate the quality of the reads and our in-house script to filter low-quality reads. The quality-passed reads were subsequently mapped to the human reference sequence (GRCh38/hg38) using the alignment tool Burrows–Wheeler Aligner (V.0.6.2).15 After removing duplicate aligned reads using SAMtools16 and recalibrating base quality scores using the Genome Analysis Toolkit (GATK),17 ,18 single nucleotide variations (SNVs) and short insertions and deletions (indels) were then identified using the GATK package and annotated by annotate variation (ANNOVAR). We included variations shared by affected cases that were not present in the control and with a read depth ≥8× and Phred score quality ≥30 for further analysis.19
Sanger sequencing
The candidate mutation identified from whole-exome sequencing was confirmed via Sanger sequencing; segregation patterns were obtained to determine whether the variant cosegregated with the scoliosis phenotype in the pedigree. Then, the variant that segregated with the clinical phenotype was screened in 503 ethnicity-matched normal controls to determine the allele frequency. PCR primers were designed using the Primer3 program (http://frodo.wi.mit.edu/). All variants identified were validated by independent PCR amplification and DNA bidirectional sequencing performed on an ABI 3130 DNA analyser.
RNA extraction and real-time QPCR
Total RNA was isolated from Epstein–Barr virus-transformed B cell lines using the RNeasy Plus mini kit (Qiagen, Hilden, Germany). cDNA was synthesised from a total of 2 µg of RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, Massachusetts, USA) and oligo (dT) primers. Reactions were carried out in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, California, USA) using Maxima SYBR Green qPCR Master Mix (Thermo Scientific, Waltham, Massachusetts, USA). The data were analysed using Bio-Rad CFX Manager Software. The mRNA expression levels of AKAP2 were analysed with one pair of primers, and the amplified products covered the location of the mutation. Expression levels were normalised to beta-actin (ACTB) (β-actin). Specific primers for each gene were as follows: AKAP2 (forward primer, 5′-AGCTTGCAGCCTGACTTAGC; reverse primer, 5′-CATTCTCTCGCGCCTTTTAG; product 117 bp) and ACTB (forward primer, 5′-CACGATGGAGGGGCCGGACTCATC; reverse primer, 5′-TAAAGACCTCTATGCCAACACAGT; product366 bp). Each assay was performed in five independent tests. The data were analysed by unpaired two-tailed t tests using GraphPad Prism V.5 software (V.5.0).
Results
Clinical characterisations
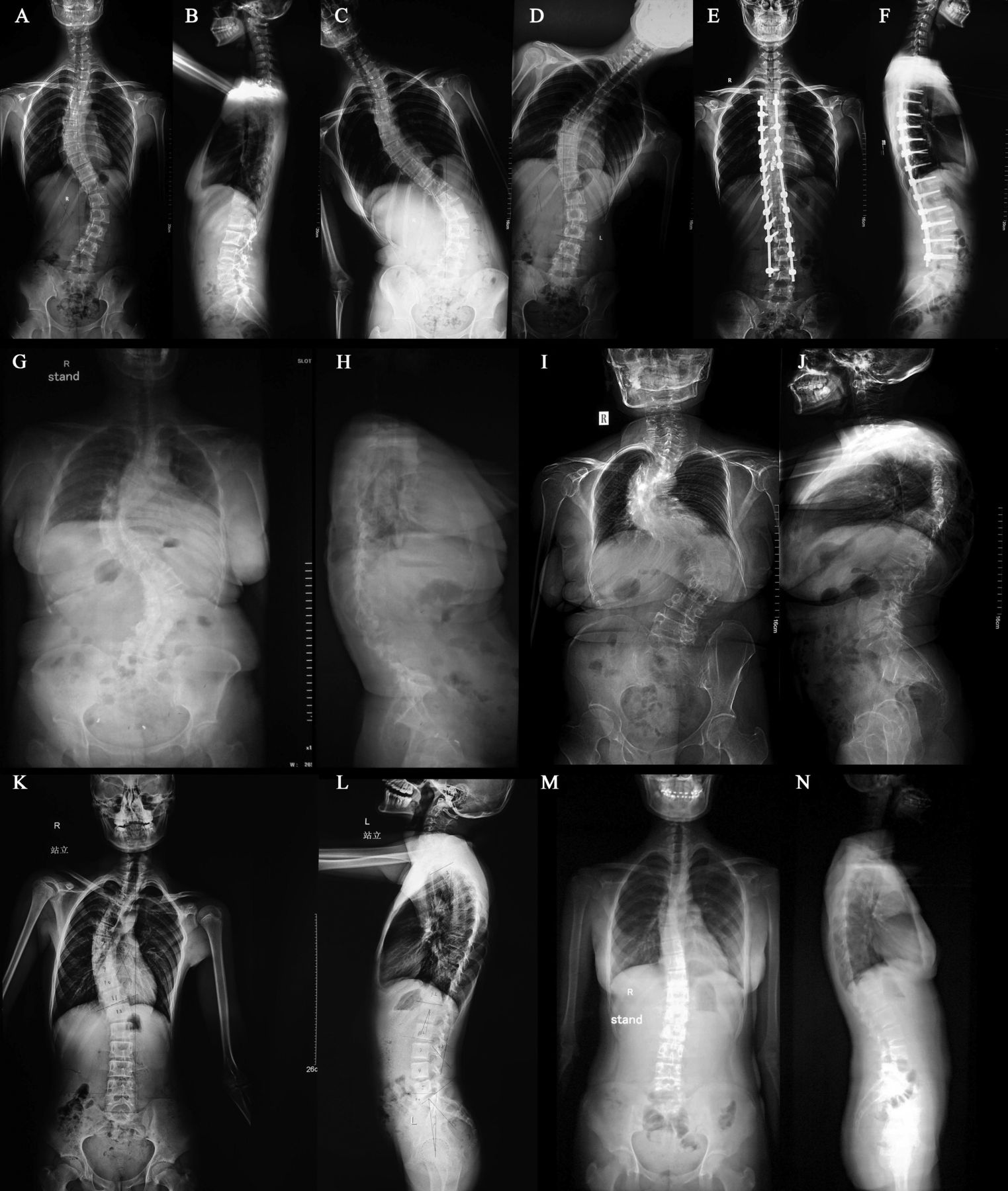
Patients distributed over four generations exhibited an autosomal-dominant mode of inheritance of AIS (figure 1A). Notably, the female members were predominantly affected, that is, there was a sex bias (four females vs one male), and females presented with a more severe phenotype than males. The proband with a 62° Cobb angle main curve underwent a corrective operation for AIS in a Chinese teaching hospital in 2006 (figure 2). By taking a careful family history, the pedigree with AIS was determined. Five individuals (II5, III7, IV9, IV10 and IV11) in this pedigree were diagnosed with AIS (figure 2). The 503 controls were ethnically matched with the patients and skeletally mature at the time of the study. Individuals with any potential evidence of skeletal diseases, metabolic disorders, growth anomalies or other diseases known to affect normal skeletal metabolism were excluded.

The AKAP2 p.E882A variant identified in the adolescent idiopathic scoliosis (AIS) family. (A) Pedigree and segregation pattern in the AIS family (asterisks indicate genomic DNA available in this study). (B) Sequence of the AKAP2 c.2645A>C variant. (C) Evolutionary conservation of the p.E882 residue (asterisks, colons and full stops above the sequences represent identity, high conservation and conservation of the amino acids, respectively). (D) RNA expression of AKAP2 in affected individuals and controls. Mean expression (±SEM) of AKAP2 in affected individuals (n=2) and controls (n=4) measured by real-time PCR.
{kind=link}
{kind=link}
(A–F) The proband (IV9), a 17-year-old girl who had adolescent idiopathic scoliosis for 4 years, and the aggravated deformity. (A and B) Preoperative anteroposterior and lateral views show a thoracic main curve of 62°, lumbar curve of 42° and thoracic kyphosis of 10°. (C and D) Preoperative anteroposterior bending views show the flexibility of the scoliosis. (E and F) Postoperative anteroposterior and lateral views show an excellent correction of the patient, where the main thoracic curve was reduced to 3°, the lumbar curve was 0° and the thoracic kyphosis was increased to 25°. (G and H) III7, a 44-year-old female who had a more than 30-year history of spinal deformity. Anteroposterior and lateral views show a thoracic main curve of 92°, kyphosis of 5° and a lumbar curve of 85°. (I and J) II 5, a 65-year-old female who had spinal deformity for over 50 years. Anteroposterior and lateral views show a proximal thoracic curve of 35°, a thoracic main curve of 102°, kyphosis of 55° and a lumbar curve of 80°. (K and L) IV10, a 10-year-old boy who was diagnosed with a spinal deformity just 1 year earlier. Anteroposterior and lateral views show a thoracic main curve of 45° and kyphosis of 25°. (M and N) IV11, a 15-year-old girl who was first identified as having scoliosis in this imaging examination. Anteroposterior and lateral views show a thoracic main curve of 22° and kyphosis of 18°.
A segregating mutation was identified in AKAP2
We generated an average of 10 Gbp of sequences with more than 77× coverage for each subject. More than 97% of the targeted bases were covered to identify SNVs and indels (see online supplementary table S1). Finally, we identified 22 370 coding variants including 11 128 non-synonymous SNVs, splicing SNVs and indels for III6; 22 505 coding variants including 11 195 non-synonymous SNVs, splicing SNVs and indels for IV10; and 22 428 coding variants including 11 098 non-synonymous SNVs, splicing SNVs and indels for IV11 (table 1).
Filtering procedures and statistics for the SNVs and indels called from the exome sequencing data
Supplementary tables
No variant in POC5 was found in our exome sequencing data. We used the following steps to filter the variants: (1) exclude high frequency SNPs (minor allele frequency (MAF)>0.01) in the 1000 genome (2015 Aug), and ESP6500 (version esp6500siv2) and ExAC (version ExAC03) databases; (2) exclude homozygous variants; (3) extract the segregating variants in the exome sequencing of individuals; (4) exclude variants in 236 in-house controls; and (5) extract variants in the reported linkage intervals (table 1, see online supplementary table S2). After this filtering analysis, three variants in MDGA1(6p21.2), AKAP2(9q31.3) and C17orf75(17q11.2) genes remained for further analysis (table 1). Subsequently, Sanger sequencing demonstrated that only one variant within AKAP2 (chr9:112810878-112934791, ENST00000434623), c.2645A>C (p.E882A), cosegregated with the phenotype in all of the family members (figure 1A, B). We also conducted segregation analyses of the variants located outside of the reported linkage regions. No other variant segregated with the AIS phenotypes in this family. This mutation was absent in the 503 population-matched normal controls and the dbSNP144, 1000G and ESP6500 databases.
The segregating mutation increased the mRNA expression level of AKAP2
Comparing the amino acid sequences of AKAP2 in multiple species revealed that the variant c.2645A>C (p.E882A) is highly conserved throughout evolution (figure 1C). Functional prediction with four programmes (sorts intolerant from tolerant (SIFT), PolyPhen2, MutationTaster and likelihood ratio test (LRT)) determined that this variant is damaging (table 2). Splice Site Prediction on this mutation with HSF 3.0 (http://www.umd.be/HSF3/HSF.html) showed ‘alteration of an exonic ESE site. Potential alteration of splicing’. Considering the hypothesis that this missense mutation influences the mRNA expression level of AKAP2, we performed QPCR using lymphocyte cells from two cases (III:7 and IV10), two controls (III:2 and III:3) within the family and two controls outside the family. The expression level of family control (III3) is regarded as ‘1’ for normalisation. The results revealed that the level of AKAP2 mRNA expression was increased significantly compared with the controls (p<0.0001) (figure 1D).
Functional prediction and conservation analysis for the cosegregating mutation
Discussion
This research identified a novel variant, c.2645A>C (p.E882A), within the AKAP2 gene that segregated with the phenotypes in the pedigree. This mutation was absent from the 6500 the National Heart, Lung, and Blood Institute (NHLBI) exome data, 1000 genomes, dbSNP144 and 503 population-matched normal controls. Functional and conservation analysis revealed that this variant is deleterious and highly conserved. The QPCR analysis showed that the expression level of mutated AKAP2 was increased significantly compared with the four controls, indicating the consequence of this mutation.
The AKAP2 gene encodes A-kinase (PRKA) anchor protein 2, which interacts with the regulatory RII subunits of protein kinase A (PKA). The AKAP2 protein most likely participates in forming polarity in signalling pathways or in constructing PKA–RII effector complexes that capture, amplify and focus diffuse transcellular cAMP signalling systems. This gene belongs to the A-kinase anchor protein (AKAP) family, whose function is to mediate PKA anchoring and binding. The specific function of PKA is localised to various subcellular locations through an interaction with AKAPs. To date, the available information on this gene and its protein is limited. The novel missense variant (c.2645A>C p.E882A) is in the highly conserved C-terminal domain (799–935 aa) of AKAP2. AKAP2 is thought to bind and position PKA, and the E882A mutation is predicted to disrupt this binding. In addition, this mutation results in a significant and extreme increase in the expression level of the mutant protein in affected individuals.
Several lines of evidence support the pathogenicity of the mutation. AKAP2 on 9q31.3 was precisely mapped to a previously reported AIS locus (IS4) on chromosome 9q31.2–q34.2. Ocaka et al11 studied a large family of British descent with autosomal dominant (AD) AIS. In this family, the proband bore a scoliotic curve of 56° determined by the Cobb method before corrective surgery, with the curvature in other affected family members ranging from 15° to 65°. The author performed a genome-wide linkage analysis in the family and obtained a maximum lod score of Zmax=3.64(θ=0.0) at marker D9S2157. Haplotype analysis identified a 21 Mb critical interval on chromosome 9q31.2–q34.2. Seven additional families showed cosegregation of the disease with marker D9S2157, and among all of the families, individuals with the disorder existed in each generation without skipping generations. Both males and females were affected, and in seven families, male-to-male transmission appeared eight times. Therefore, transmission of AIS is compatible with an AD inheritance pattern. The family we studied has much in common with these families, such as the observation of multigenerational disease, the observation of members affected with an AD mode of AIS, and the observation of affected subjects with a more severe spinal curvature.
The AKAP2 gene was previously reported to be interrupted by a translocation in a case with Kallmann syndrome and bone anomalies.20 The patient had a kyphotic deformity, flatfoot with the absence of the fifth toe bilaterally, eversion of the foot, abnormal fusion of the scaphoid with the astragalus bone and dysplasia of the hip. To explore how AKAP2 contributes to the disorder, the author detected mAkap2 expression in mouse embryos by in situ hybridisation studies. A high level of expression in cartilaginous structures of the embryo was observed, in correspondence with the phenotype of the proband carrying the translocation in this study. These studies revealed that Akap2 contributes to the development of cartilage. The abnormal response of growth plate chondrocytes to the action of melatonin in both proliferation and differentiation might be associated with abnormal endochondral ossification.21 Magnetic resonance images and histomorphometric studies have disclosed that abnormal endochondral ossification affects skeletal growth in patients with AIS22–24 and is considered to be a promoting factor in the pathogenesis of the disease. Moreover, it has been pointed out that the PKA pathway in osteocytes is to stimulate bone growth and mediate the anabolic skeletal response.25 Therefore, we have reason to propose that AKAP2 plays an important role in the pathogenesis of AIS.
In conclusion, our study first indicated AKAP2 as a novel gene mutated in the AIS family by combining exome sequencing with segregation analysis, conservation analysis and expression analysis. Further cell functional studies and animal studies of AKAP2 will be indispensable to interpret its role in the aetiopathogenesis of AIS.
Acknowledgments
We are particularly grateful to all the patients and their family members who participated in this study.
References
Footnotes
Contributors All authors have read and approved the final manuscript. Study design: ZH, BW and XL. Clinical investigations and sample collection: WL, YWL and YD. Performing the experiments: WL, LZ, DT, YL, YP and KX. Data interpretation and analysis: WL, HG and YZ. Drafting the manuscript: WL, YWL and HG.
Funding The research was supported by the National Natural Science Foundation of China (81371919, 31400919 and 81330027), the National Basic Research Program of China (2012CB517902) and the Natural Science Foundation of Hunan Province (2015JJ2160).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study was approved by the Institutional Review Board of The State Key Laboratory of Medical Genetics and adhered to the tenets of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.