Article Text

Abstract
Background Mutations in known genes for inherited forms of Parkinson's disease (PD) account for <30% of familial PD (FPD) implying that more causal gene(s) remain to be identified. We attempted to discover the putative causal variant in an Indian family with autosomal-recessive juvenile Parkinsonism (ARJP), tested negative for mutations in PARK2, PINK1 and DJ1.
Methods Whole exomes of two affected siblings were sequenced. Variants prioritised were screened for segregation with disease in the family by targeted sequencing. Gene thus identified was screened for index/additional exonic mutations, if any, in an independent PD cohort by PCR sequencing. Variants observed were functionally validated in differentiated PC12 cells.
Results A novel homozygous frameshift mutation, c.89_90insGTCGCCCC in exon 1 of podocalyxin-like gene (PODXL, 7q32–33), resulting in loss of protein, segregated with disease in the family. Mutant allele was absent in 186 healthy controls screened by PCR sequencing and in control exomes available in the laboratory and public databases. Screening of additional 212 sporadic and 68 FPD cases identified three novel heterozygous missense variants namely c.1285C>A, c.1118G>A and c.881G>A in three unrelated cases. Significant differences in neurite branching and length (p<0.0001) were observed in PC12 cells with wild-type and mutant constructs.
Conclusions Based on the genetic and functional evidence in this study and literature support on the role of PODXL in neural development, a novel frameshift mutation in PODXL seems to be the likely cause of ARJP in this family. This is the first report suggesting the possible role of a neurodevelopmental pathway in PD aetiology.
- Parkinson-s disease
- Clinical genetics
- Genetics
- Neurosciences
- Genome-wide
Statistics from Altmetric.com
Introduction
Selective degeneration of dopaminergic neurons in the substantia nigra pars compacta of the midbrain is the primary pathology underlying Parkinson's disease (PD), the second most common neurodegenerative disorder. Despite intense research efforts, the exact mechanism of this degeneration, which occurs mostly in a sporadic manner in the elderly population, remains unknown. Xenobiotics were believed to be the primary contributors to this most common form of movement disorder,1 but a genetic basis with several candidate genes has since been shown to be associated with PD.2 ,3 These genes come from a number of pathways including the dopaminergic pathway, ubiquitin proteasome system, synaptic homeostasis, autophagy, lysosomal pathway, mitochondrial maintenance, axonal transport, programmed cell death and inflammatory pathways, all of which have been implicated to lead to neuronal cell death.4 ,5 Unlike a polygenic contribution from these pathways to the common sporadic forms of PD, mutation(s) in single genes are known to be causal in Mendelian forms of the disease, which constitute ∼10% of all PD cases. Genome-wide linkage scans in well-characterised PD families were successful in discovering six genes namely SNCA, PARK2, PINK1, DJ1, LRRK2 and EIF4G1.6 ,7 More recently, next-generation sequencing (NGS) technology has helped to identify three more genes namely VPS35,8 DNAJC139 and CHCHD2.10 Though the contribution of DNAJC13 to familial and sporadic PD across different populations remains to be investigated, mutations in the other eight genes account for <30% of all familial forms of the disease examined to date,7 ,11 though with varied frequency across ethnic groups. For example, mutations in LRRK2 were seen in 30%–42% and 2.6% of autosomal-dominant PD cases from Tunisia and USA, respectively,12 but 0.3% in Japanese13 and <0.1% in North Indian PD cases.14 On the other hand, mutations in PARK2 explain up to 50% familial PD (FPD) cases and ∼15% sporadic PD cases globally,15 but are seen in ∼14.3% of FPD and 6.9% in young-onset and 5.9% in late-onset sporadic cases from India.16 It is also noteworthy that almost all these FPD genes known to date are involved only in the ubiquitin proteasomal degradation pathway, mitochondrial maintenance or autophagy-lysosome pathway.4 It is very likely that novel mutations in more genes from these and other known or yet unknown pathways remain to be identified to fully understand PD biology. Analysis of FPD cases continues to be one of the major approaches for discovery of additional causal variants, and the power of the recent NGS tools, irrespective of family size, makes this an attractive strategy.
With this background, we carried out exome sequencing of a 10-member Indian family with autosomal-recessive juvenile Parkinsonism (ARJP) and which was negative for mutations in three known ARJP genes namely PARK2, PINK1 and DJ1. We identified a novel, putative disease-causing variant in podocalyxin-like gene (PODXL), coding for a neural adhesion molecule, and not implicated in PD aetiology to date.
Methods
Recruitment of study subjects
A 10-member ARJP family (figure 1) was recruited from Aligarh, Uttar Pradesh, India by clinicians KP and MB. PD was diagnosed using the UK PD Brain Bank clinical diagnostic criteria.17 There was a family history of consanguinity, though the parents (I1, I2, figure 1) and five remaining siblings (II1, II2, II6, II7 and II8, figure 1) were unaffected. A detailed description of the clinical features of all three affected siblings is presented in table 1. Following clearance from ethical committees of participating institutions, informed consent and 10 mL peripheral venous blood was obtained from each of the study subjects.
Clinical characteristics of affected individuals in the study family with ARJP

Pedigree of an Indian family with autosomal-recessive juvenile Parkinsonism. I1, I2 unaffected parents; II3, II4, II5 affected siblings; II1, II2, II6, II7, II8 unaffected siblings; * exome sequenced.
Genetic analysis
Genomic DNA was isolated using the conventional phenol chloroform extraction method used in the laboratory. Mutation screening in all exons of PARK2, PINK1 and DJ1 was carried out by PCR sequencing using primers described elsewhere16 and listed in the online supplementary tables S1 and S2.
NGS and data analysis
Exome sequencing of two (II4, II5, figure 1) of the three affected siblings aged 16 and 13 years at disease onset, respectively, from the family under study was carried out. Library preparation and enrichment using NimbleGen V.3 kit and sequencing on Illumina HiSeq2000 were done using a commercial facility (Axeq Technologies, USA). Raw data obtained were subjected to stringent pre-alignment quality control (QC) for quality-based filtering using FASTX-Toolkit,18 alignment by BWA19 and post-alignment QC with SAMtools.20 Target coverage and depth (see online supplementary table S3) were assessed by BEDTools,21 followed by variant-calling using SAMtools,20 and GATK;22 and annotation using wANNOVAR.23
All SNPs with minor allele frequency (MAF) >0.01 reported in databases such as dbSNP135, 137 and 138,24 1000 genomes25 and NHBLI 6500 exomes26 were excluded to start with. Keeping in mind the autosomal-recessive inheritance pattern, homozygous exonic variants shared between the two affected individuals were prioritised. In parallel, KGGseq27 was also used to filter variants (see online supplementary table S4).
Prioritised variants (see online supplementary table S5) were then checked for segregation with the disease in the remaining family members by targeted sequencing using HaloPlex enrichment kit (Agilent, USA) on Illumina HiSeq2500 using a commercial facility (Strand Genomics, India). Index mutation(s) were screened in unrelated control individuals (n=186) by PCR-Sanger sequencing; and checked in 144 healthy control exomes and 74 non-PD exomes (of subjects with other disorders), available in the laboratory.
Further, index mutation(s) and additional exonic variants, if any, in the gene(s) segregating with the disease phenotype were screened in a PD cohort (n=212 sporadic and 68 familial cases) available in the laboratory (see online supplementary table S6 for primer details) by PCR-Sanger sequencing. All the variant(s) identified as above were then taken forward for suitable functional studies in vitro.
In vitro functional analysis
Transfection and generation of stable PC12 cells
Stably transfected PC1228 cells were generated with the wild-type (WT) and mutant open reading frame (ORF) constructs of the gene of interest using a standard protocol. Briefly, TrueORFGold expression-validated ORF clone of gene of interest with a C-terminal myc-DDK-tag was obtained in a pCMV6 vector (RC210816, OriGene Technologies, Rockville, Maryland, USA). Site-directed mutagenesis to generate the mutant(s) (see online supplementary table S7 for primer details) was performed as per Stratagene Quikchange II XL kit-protocol from this WT ORF along with subsequent transformation of XL10Gold Escherichia coli cells followed by confirmation of the mutation(s) by Sanger sequencing. Stable PC12 cells for each of the constructs were generated by transfection-mediation using Lipofectamine LTX and Plus reagent (Thermo Fisher Scientific) and G418 selection. Transfections and subsequent stable-line generations were done twice per construct to obtain biological replicates. Untransfected (UT) PC12 cells were used as controls.
Protein expression confirmation
Expression of WT and mutant proteins from the respective stable PC12 cells was confirmed by western blot analysis using standard protocols. Briefly, equal quantities of total cell lysates prepared from the respective stable lines, after protein estimation, were run on 12% sodium dodecyl sulfate-polyacrylamide gels, blotted and probed with appropriate antibodies. β-actin was used as an internal control. Antibodies used were mouse monoclonal anti-DDK-tag (TA50011-100, OriGene Technologies) diluted to 1:2000; rabbit polyclonal anti-β-actin (ab8227, Abcam) diluted to 1:2000; goat anti-mouse IgG–horseradish peroxidase (HRP) conjugate (32430, Thermo Fisher Scientific) diluted to 1:5000; and goat anti-rabbit IgG–HRP conjugate (ab97200, Abcam) diluted to 1:10 000.
Neurite branching assay
Eight thousand to 10 000 cells from each of the stable PC12 lines were seeded per chamber on collagen-coated four-well chamber slides (SPL Lifesciences, Korea) and grown for 24 h in 1× Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% horse serum, 5% fetal bovine serum and additional supplements. UT PC12 cells were used as controls. After 24 h, medium was changed to reduced-serum differentiation medium (1× DMEM+1% horse serum+100 mg/mL nerve growth factor). Differentiation medium was changed every 48 h. Cells were allowed to differentiate for 4 days, and then permeabilised and fixed on day 5 with methanol. Slides were then prepared for immunofluorescence by treating with primary (mouse monoclonal anti-DDK-tag, TA50011-100 OriGene Technologies, diluted to 1:1000) and secondary (donkey anti-mouse IgG-AlexaFluor594, 715-585-151, Jackson ImmunoResearch, diluted to 1:500) antibodies and visualised on Olympus BX51 fluorescence microscope. Images captured on 20× were then analysed using IMAGEJ2 software29 for two parameters, namely total neurite length per cell and total number of branch points per cell. At least 80 cells per category were analysed for both these parameters. Mann–Whitney test was performed to generate p values between WT and each of the mutants and p values <0.05 were considered to be significantly different. All statistical analyses were done using GraphPad Prism trial V.6.01 for Windows, GraphPad software, La Jolla, California, USA.
Results
Novel frameshift mutation in PODXL identified as a potential causal variant in the family with ARJP
The 10-member family with two unaffected parents, three affected and five unaffected children included in this study were screened and found to be negative for mutations in PARK2, PINK1 and DJ1. The oldest affected sibling (II3, figure 1) died after recruitment but prior to exome sequencing in this family (at the age of 22 years). Exome sequencing of the two younger affected brothers (aged 16 and 13 years at disease onset) yielded total target coverage (>10×) of 93.9% and 72.9×, and 64.3× mean target depth, respectively (see online supplementary table S3). Downstream analysis with optimal stringency and removal of shared variants with frequencies >0.01 from different databases (as detailed in ‘Methods’ section) was done, which yielded 17 homozygous shared exonic variants in 17 different genes (see online supplementary table S5). On segregation analysis of these variants in the remaining family members by targeted sequencing using a customised HaloPlex enrichment kit (Agilent), a single novel, homozygous frameshift mutation in PODXL was found to be shared among all three affected siblings and thus segregating with the disease in this family (figure 2A, B). Both parents (I1, I2, figure 1) were found to be heterozygous for this change, and of the remaining five unaffected children, three (II1, II2 and II7) were heterozygous for this index mutation, and two (II6, II8) had homozygous WT alleles (figure 2A). This mutation, a tandem repeat expansion of the six bp motif namely GTCGCC followed by insertion CC (c.89_90insGTCGCCCC, nomenclature as per Human Genome Variation Society (HGVS) convention), causes a frameshift immediately after a proline–serine stretch at Ser31, in the WT protein. While the proline–serine expansion (n=4) observed in affected individuals is a reported polymorphism (rs759639123, MAF=0.3), the frameshift insertion in exon 1 of the gene leads to a stop codon gain at codon 168 of the 558 (NM_001018111.2, isoform 1) or 526 (NM_005397.3, isoform 2) codon transcripts from the gene (see online supplementary figure S1). This change in the mRNA would trigger nuclear nonsense-mediated decay mechanism leading to absence of protein in the affected members with the homozygous variant. PODXL, or podocalyxin-like, located on chromosome 7q32–33 (with nine exons and two isoforms), is a known sialomucin, expressed largely in the brain, heart and kidneys.30 There are 136 organisms having orthologs with this gene. Loss of function of the mouse ortholog of PODXL has been previously reported to distort neuronal branching,31 which may have debilitating implications in neuronal development and neurodegenerative diseases. Thus, the observed novel mutation in PODXL may be potentially causing ARJP in the family under study.
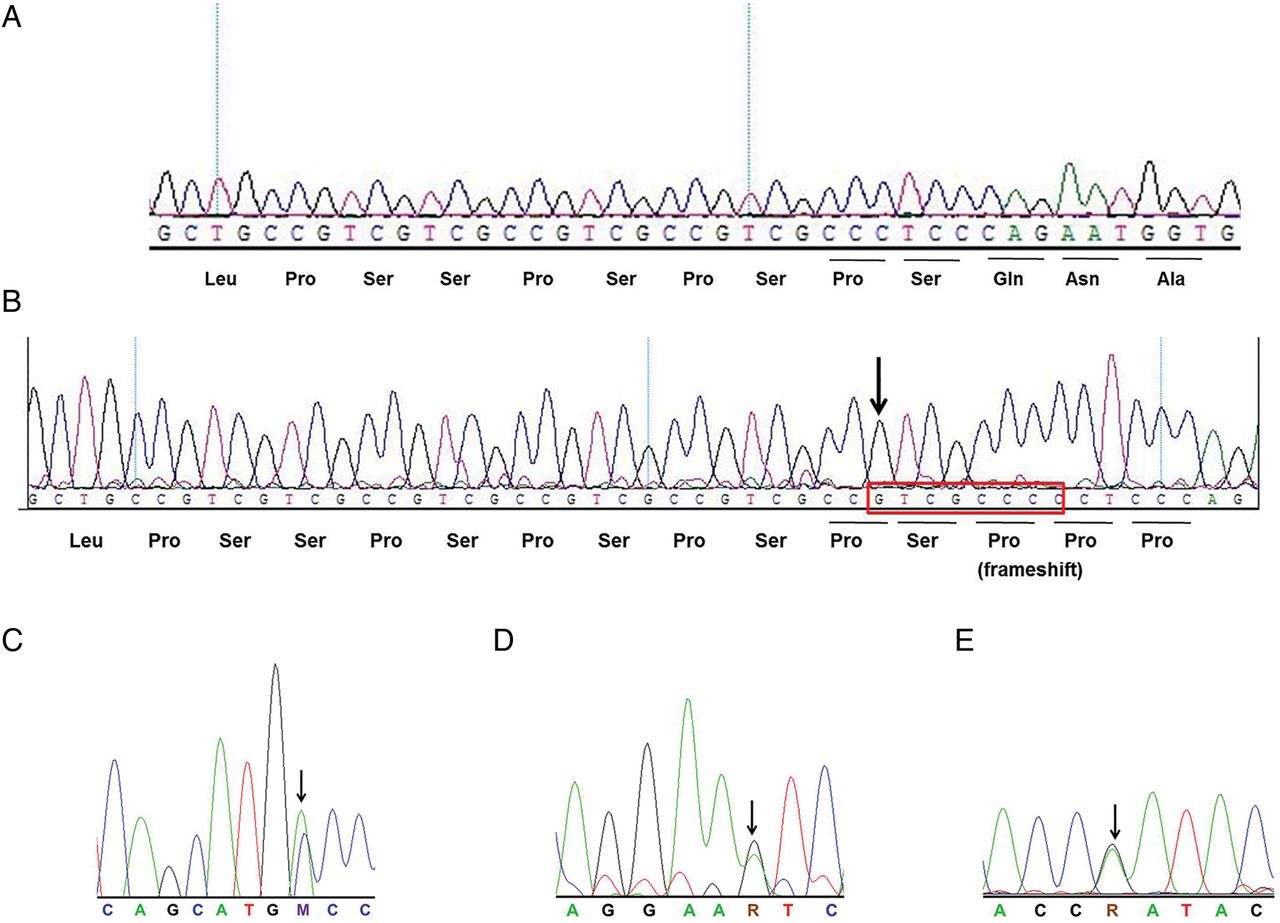
Electropherograms depicting wild-type (WT)/mutant sequences in podocalyxin-like (PODXL). (A) Homozygous WT sequence found in two unaffected siblings and unrelated controls. (B) Homozygous frameshift mutation (c.89_90insGTCGCCCC) found in three affected individuals; arrow indicates site of frameshift; red box indicates insertion sequence; (C) c.1285C>A, p.P429T denoted by ‘S’ in a young-onset Parkinson's disease case; (D) c.1118G>A, p.S373N denoted by ‘R’ in a late-onset PD case; and (E) c.881G>A, p.R294Q denoted by ‘R’ in a familial Parkinson's disease case; arrows indicate sites of mutations.
Additional heterozygous mutations in PODXL in an unrelated PD cohort
On screening all eight coding exons of PODXL by PCR sequencing in 280 PD cases from the PD DNA bank available in the laboratory, we identified three novel heterozygous missense mutations. Mutation NM_005397.3:c.1285C>A:p.P429T (P429T) in exon 7 (figure 2C) was found in a young-onset PD (YOPD) male with age at disease onset (AAO) of 22 years and age at time of examination (AAE) of 25 years. Mutation NM_005397.3:c.1118G>A:p.S373N (S373N) in exon 5 (figure 2D) was found in a late-onset PD (LOPD) male with AAO of 71 years and AAE of 73 years. Mutation NM_005397.3:c.881G>A:p.R294Q (R294Q) in exon 3 (figure 2E) was observed in an FPD male with AAO of 42 years and AAE of 62 years. This FPD subject has an affected maternal uncle (deceased) and an affected maternal first cousin. All three unrelated heterozygous carriers of these novel mutations are from North India and have classical PD symptoms. They have also been previously screened and found negative for mutations in PARK2,16 PINK1 and DJ1 (unpublished data). The three heterozygous mutations and the index mutation were absent in 186 age-matched unrelated control individuals screened by PCR-Sanger sequencing, 144 unrelated healthy control exomes and 74 non-PD exomes (of subjects with other disorders) available in the laboratory. These variants have also not been reported in over 60 000 unrelated non-PD exomes from Exome Aggregation Consortium (ExAC) database.32 All the four mutations (SCV000258390, SCV000258391, SCV000258392 and SCV000258393) are in exons present in both isoforms of PODXL (see online supplementary figure S1). In silico prediction using SIFT,33 PolyPhen234 and MutationTaster35 suggested the likely pathogenic nature of the three non-synonymous mutations identified.
Novel mutations in PODXL alter neurite branching pattern in PC12 cells differentiated into neurons
The homozygous frameshift mutation in the family under study, which would lead to a total loss of protein, as described above, was not analysed further. PODXL is mostly expressed in the brain, heart and kidney. Due to non-availability of either the tissue samples from the affected individuals or the autopsy samples from the deceased affected sibling, absence of protein in these affected individuals could not be demonstrated. Further, it may be noted that loss of protein cannot be investigated in an overexpression model due to endogenous PODXL expression but a knock-out strategy would be useful. However, such a knock-out mouse model showing increased neurite outgrowth and branching has already been reported by another group.31 This evidence lends substantial support for the functional significance of this protein. Based on this knowledge, the functional significance of the three additional novel mutations identified in our PD cohort was investigated in vitro using the neurite branching assay. This was done by first generating stable lines of the WT PODXL ORF and three mutant ORFs, namely P429T, S373N and R294Q, in PC12 cells after protein expression in all the lines was confirmed by western blotting (figure 3). After 5 days of differentiation into neurons, the neurite branching profile, scored for total neurite length and total number of branch points per cell, was compared between the WT and each of the three mutants (figure 4). Mutant R294Q (observed in FPD) showed a significant increase (Mann–Whitney p<0.0001) in both total length of neurites per cell and in the total number of branch points per cell. Mutants P429T and S373N showed a significant increase (Mann–Whitney p<0.0001) in the total number of branch points per cell, but no significant change in total neurite length per cell (figure 4). There was no significant difference between WT and UT control cells scored for the same parameters, which can be explained by the endogenous expression of PODXL in the rat cells. These findings are suggestive of the functional significance of the mutations in PODXL.

Western blot profile confirming protein expression. (A) Podocalyxin-like (PODXL)-myc-DDK expression in wild-type (WT) and mutant lines P429T, S373N and R294Q; (B) β-actin expression across untransfected (UT), WT, P429T, S373N and R294Q lines.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neurite branching profile in wild-type (WT) and mutant stable PC12 cells differentiated into neurons. (A–D) Representative images captured under 20× objective, on Olympus BX51 fluorescence microscope. Stable PC12 lines expressing podocalyxin-like (PODXL)-myc-DDK: (A) WT. (B–D) Mutants P429T, S373N and R294Q, respectively. (E and F) Median (denoted by short horizontal lines for each group) with aligned dot plots. (E) Total neurite length per cell. (F) Total number of branch points per cell.
Discussion
Mendelian forms of PD have undoubtedly a major genetic component compared with the predominant late-onset sporadic PD cases.36 Notable early efforts in PD gene discovery using genome-wide linkage scans identified three genes, namely PARK2, PINK1 and DJ1, for ARJP. These have been extensively investigated across ethnic groups. However, mutations in these genes explain <15% of ARJP cases in India,16 ,37 ,38 and ∼1%–50% of ARJP cases and ∼1%–20% of YOPD cases in Caucasians15 implying that more disease-causing genes remain to be discovered. This is not unexpected, considering that PD is a genetically and clinically heterogeneous condition and genes from a range of pathways could contribute to the etiopathology of this neurodegenerative condition. It is at this juncture that the hypothesis-free exome sequencing approach has become a powerful tool and PD families (as well as families with other monogenic disorders), irrespective of their size, are an enviable resource to facilitate new disease-causing gene variant discovery.
Using this approach of exome sequencing in an ARJP family from North India, we identified a novel putative disease-causing variant (c.89_90insGTCGCCCC) in PODXL, a gene whose contribution to PD is hitherto unreported. As already mentioned in the ‘Results’ section, the index mutation led to loss of protein and, therefore, could not be validated in this overexpression study in PC12 cells differentiated into neurons. However, as mentioned earlier, loss of function of the mouse ortholog, in a knock-out study reported previously, has shown increased neurite outgrowth and branching.31 This may be considered as evidence for the role of PODXL in the pathology observed in the study family.
PODXL is a highly sialylated adhesion glycoprotein, abundantly expressed in the brain, second only to the neural cell adhesion molecule (NCAM).31 Several studies have demonstrated the role NCAM plays in modulating neural plasticity and development.39–41 NCAM bound to polysialic acid (PSA) is known to regulate axonal fasciculation42 and synaptic stablilisation.43 To some extent, an early in vivo study on PODXL-deficient mice31 has been insightful in understanding whether sialylated and non-sialylated forms of neural PODXL play a similar role to PSA-NCAM in neural development. The authors were able to show that though PSA-bound forms of PODXL were an essential negative regulator of neurite outgrowth, similar to PSA-NCAM, non-sialylated PODXL was required for regulation of neurite branching. They reported increased neurite outgrowth and branching, and a reduction in the number of synapses formed in hippocampal and cortical neurons isolated from brains of these PODXL-deficient mice, suggesting this protein to be a negative regulator of neurite outgrowth and branching, with a direct involvement in synaptogenesis. The same study demonstrated interactions between PODXL from brains of mice, and sodium–hydrogen exchanger regulatory factors 1,2 (NHERF1,2), Ezrin and G-coupled protein and GTPase Rho A,G, thereby proposing that negative regulation of neurite outgrowth and branching was due to PODXL-mediated activation of NHERF1,2/Ezrin/RhoA,G complex. Independently, in another study, MRI of brains in PODXL-deficient mice has indicated a potential role of this protein in neuronal development.44 In the same study, the absence of PODXL in the aetiology of schizophrenia and other neurodegenerative diseases has also been discussed. A significant ventricular enlargement was reported to correlate with the ablation of PODXL in mouse brains. In other studies, such ventricular enlargement was seen to correspond with cognitive decline,45 and motor asymmetry46 in PD cases, which they believed may serve as a marker for neurodegenerative disease progression. Interestingly, a reduction in sialic acid and sialylated membrane-bound molecules has been reported to trigger inflammatory responses in the brain by microglial activation.47 The downstream neurotoxic effects of such microglia-mediated inflammatory pathways have been implicated as one of the mechanisms of PD pathogenesis.47 Therefore, it may not be unexpected, that a loss of PODXL, a polysialylated cell adhesion molecule, might lead to PD through a microglia-activated immune response. These speculations are amply corroborated by our own findings of a frameshift mutation leading to loss of protein in three affected siblings, all of whom had a debilitating form of juvenile-onset PD, including the reported death of the oldest sibling during the study period.
On the other hand, on screening for the index mutation and/or additional variants in PODXL, we found three novel, although heterozygous, mutations one each in YOPD, LOPD and FPD cases whose ages at onset were considerably later than in the index family. These mutations were predicted to be functionally relevant using in silico tools. The experimental observations of alteration in neurite branching pattern, either in the total neurite length or total number of branch points or both (figure 4), support the in silico predictions. Though these findings are promising, the mechanism underlying PODXL variants influencing neurite branching is not yet well understood. In this study, we have been able to compare the neurite branching pattern of WT and mutants in vitro, but a direct correlation of the role of PODXL in PD pathogenesis remains to be demonstrated in an animal model. Therefore, we would refrain from concluding on the causality of these mutations in these three unrelated cases but may speculate that they probably contribute to the disease as risk variants in addition to other genetic and non-genetic determinants that the carriers may harbour. This may be similar to the observations of several sporadic PD cases with heterozygous mutations in PARK2.48
Finally, based on the demographic history of genetically distinct Indian populations including the recent report of Indians showing a higher average inbreeding coefficient compared with European populations,49 an increased incidence of autosomal-recessive diseases therein is highly probable. Therefore, new disease causal variant discovery efforts with contemporary deep sequencing tools in Indian populations may be immediately rewarding. In summary, with compelling genetic evidence and in vitro functional characterisation, PODXL may be considered as the fourth putative causal gene discovered for autosomal-recessive PD warranting investigation in other ethnic groups.
Acknowledgments
We are thankful to all the participants in this study. We acknowledge contributions of Ms Anjali Dhyani for genomic DNA isolation, Ms Alina Genis for mutation screening of PARK2, PINK1 and DJ1 genes, Mr Surojit Bose, LeadInvent Ltd., India for assistance in establishing a bioinformatics pipeline for data analysis, Link Biotech Ltd., India and Central instrumentation facility, UDSC for Sanger sequencing services. We gratefully acknowledge the infrastructure support provided to the Department of Genetics, UDSC, by the University Grants Commission, New Delhi under Special Assistance Programme and Department of Science and Technology, New Delhi under FIST and DU–DST PURSE programmes. We are thankful to Prof Dr Peter Heutink, German Center for Neurodegenerative Diseases (DZNE), Tubingen, Germany for critical reading of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors SS, MB, UBM, RCJ and BKT were responsible for the concept and design of the study. KP, MB and UBM were responsible for diagnosis, recruitment, clinical characterisation and blood collection for all study subjects. SS and BKT acquired, analysed and interpreted the data. SS and BKT wrote the first draft of the manuscript. All authors read the draft, provided their inputs and agreed on the final version of the manuscript before submission.
Funding This work was supported by the Department of Science and Technology, Govt. of India, New Delhi (grant number SP/SO/B-55/2000), and Department of Biotechnology, Govt. of India, New Delhi (grants number BT/PR2425/Med/13/089/2001 and number BT/PR14500/MED/12/478/2010) to BKT, MB, UBM and RCJ; and Council of Scientific and Industrial Research, New Delhi, India for JRF and SRF to SS.
Competing interests None declared.
Ethics approval Institutional Ethical Committees of AIIMS and University of Delhi South Campus.
Provenance and peer review Not commissioned; externally peer reviewed.