Article Text

Abstract
VACTERL association is a condition comprising multisystem congenital malformations, causing severe physical disability in affected individuals. It is typically defined by the concurrence of at least three of the following component features: vertebral anomalies (V), anal atresia (A), cardiac malformations (C), tracheo-oesophageal fistula (TE), renal dysplasia (R) and limb abnormalities (L). Vertebral anomaly is one of the most important and common defects that has been reported in approximately 60–95% of all VACTERL patients. Recent breakthroughs have suggested that genetic factors play an important role in VACTERL association, especially in those with vertebral phenotypes. In this review, we summarised the genetic studies of the VACTERL association, especially focusing on the genetic aetiology of patients with vertebral anomalies. Furthermore, genetic reports of other syndromes with vertebral phenotypes overlapping with VACTERL association are also included. We aim to provide a further understanding of the genetic aetiology and a better evidence for genetic diagnosis of the association and vertebral anomalies.
- VACTERL association
- Vertebral anomalies
- Gene
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Overview of VACTERL association
VACTERL association is a condition with multisystem congenital malformations: Vertebral anomalies (V), anal atresia (A), cardiac malformation (C), tracheo-oesophageal fistula (TE) with or without oesophageal atresia, renal dysplasia (R) and limb abnormalities (L).1 ,2 It was first named as VATER (without ‘C’ and ‘L’) association in 1973.3 The prevalence of VACTERL/VATER association is between 1/7000 and 1/40 000.4 ,5
As there is no available objective laboratory test for its diagnosis, VACTERL association is diagnosed totally based on the clinical manifestations mentioned above. Most clinicians and researchers require the presence of at least three component features for diagnosis. Besides, due to its heterogeneous phenotype and the abundance of overlapping defects of other syndromes, VACTERL association is typically considered a diagnosis of exclusion5–8 with no clear evidence for an alternative or overlapping diagnosis such as Coloboma, Heart anomaly, Atresia of choanae, Retardation of mental and somatic development, Genital hypoplasia, Ear abnormalities (CHARGE) syndrome, DiGeorge syndrome and Pallister–Hall syndrome. The presence of other features not typically seen in VACTERL association may suggest other disorders. Thus, a physical examination and family history are essential to rule out potentially overlapping diagnoses. It is worth mentioning that 5–10% patients with Fanconi anaemia (FA) have birth defects meeting the diagnosis of VACTERL association with hydrocephalus (VACTERL-H).9 ,10 It is suggested that FA with VACTERL-H should be treated separately from the VACTERL association because of the core characteristics of FA such as haematological anomalies and skin pigmentary changes, the different frequencies of VACTERL-associated phenotypes and the prognosis and therapeutic intervention.10 ,11
Although the clinical criteria for VACTERL association appear to be straightforward, the overlapping in either clinical manifestation or genetic finding is challenging for clinicians and geneticists. The CHD7 gene mutation, which is proved to be associated with CHARGE syndrome, may also be found in patients diagnosed with VACTERL association, even CHARGE syndrome is clinically excluded.12 Besides, most of the conditions listed are monogenic disorders. Careful genetic evaluation may help ruling out these conditions. In this review, we listed the related monogenic diseases that share two more overlapping manifestations and their genetic findings (table 1). We propose that(1) these syndromes as well as these candidate genes should be considered in diagnostic and genetic studies in VACTERL association; and (2) VACTERL syndrome remains a diagnosis of exclusion following a thoughtful clinical evaluation and consideration of genetic testing for overlapping syndromes.
Monogenic diseases overlapping with VACTERL association
Prior studies have estimated that 90% of the patients diagnosed with VACTERL association had three or fewer phenotypes (referred to as VACTERL-like association) and <1% of patients had all six anomalies.4 Although the frequency of the six clinical features (CFs) varies, vertebral anomalies is the most common observation in many cohorts of VACTERL association, which have been reported in approximately 60–95% of affected individuals.7 ,30–33 Additionally, vertebral anomalies are the most prevalent findings in the first-degree relatives of the probands in some cohorts,34 ,35 thus highlighting the importance of vertebral anomalies as a major diagnostic feature for VACTERL association. In this review, we will summarise the genetic studies of the VACTERL association with an emphasis on vertebral anomalies.
Vertebral anomalies
Vertebral anomalies in VACTERL association can be classified as (1) failure of formation, such as hemivertebrae, butterfly or wedge-shaped vertebrae; (2) failure of segmentation such as vertebral bars, fused vertebrae and block vertebrae; and (3) a combination of these two features, resulting in a mixed deformity.36 ,37 Rib anomalies such as rib fusion and increased or decreased number of ribs are commonly accompanied with vertebral anomalies. In some studies, rib anomalies may occur without vertebral anomalies.7 ,30 ,38 ,39 Although patients with anorectal malformations may be have dysplastic sacral vertebrae, it is not clear whether these should be regarded as a vertebral anomalies component for diagnosis of VACTERL syndrome.2 Clinical signs of scoliosis or kyphosis may be the first sign of vertebral anomalies when VACTERL association is suspected.40 Radiology is needed for discerning vertebral and rib anomalies.
As an example, we present a 2-year-old Chinese boy with VACTERL association. He was born with oesophageal atresia that was surgically corrected 4 days later. He had an uneventful infancy until his mother found him with a hump at lower waist a year later. Spinal X-ray and CT scan found a left hemivertebra between L3 and L4, and a right hemivertebra between L5 and S1 (figure 1), which caused evident lumbar scoliosis. He also had an extra thoracic vertebra and an extra pair of ribs without clinical symptoms. Abdominal ultrasound examination revealed horseshoe kidney without impairment of his renal function. He underwent resection of both hemivertebrae with internal fixation and recovered well postoperatively.
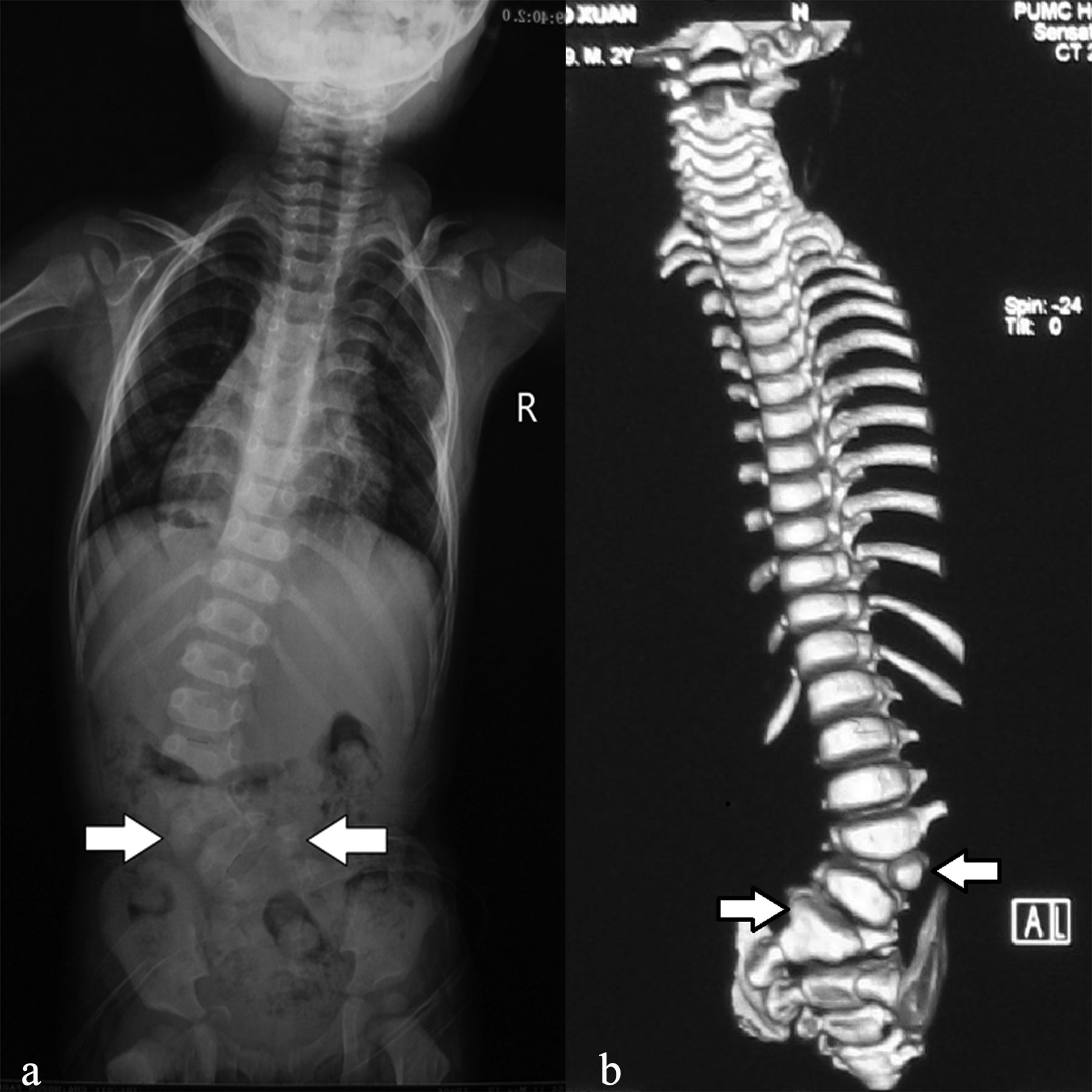
Radiology of a 2-year-old boy diagnosed with VACTERL association. Preoperative spinal X-ray (A) and CT scan (B) revealed a left hemivertebra between L3 and L4, and a right hemivertebra between L5 and S1 that was fused with S1 vertebra (white arrows). R, right side of the body; VACTERL, vertebral anomalies (V), anal atresia (A), cardiac malformations (C), tracheo-oesophageal fistula (TE), renal dysplasia (R) and limb abnormalities (L).
Genetic studies on VACTERL association
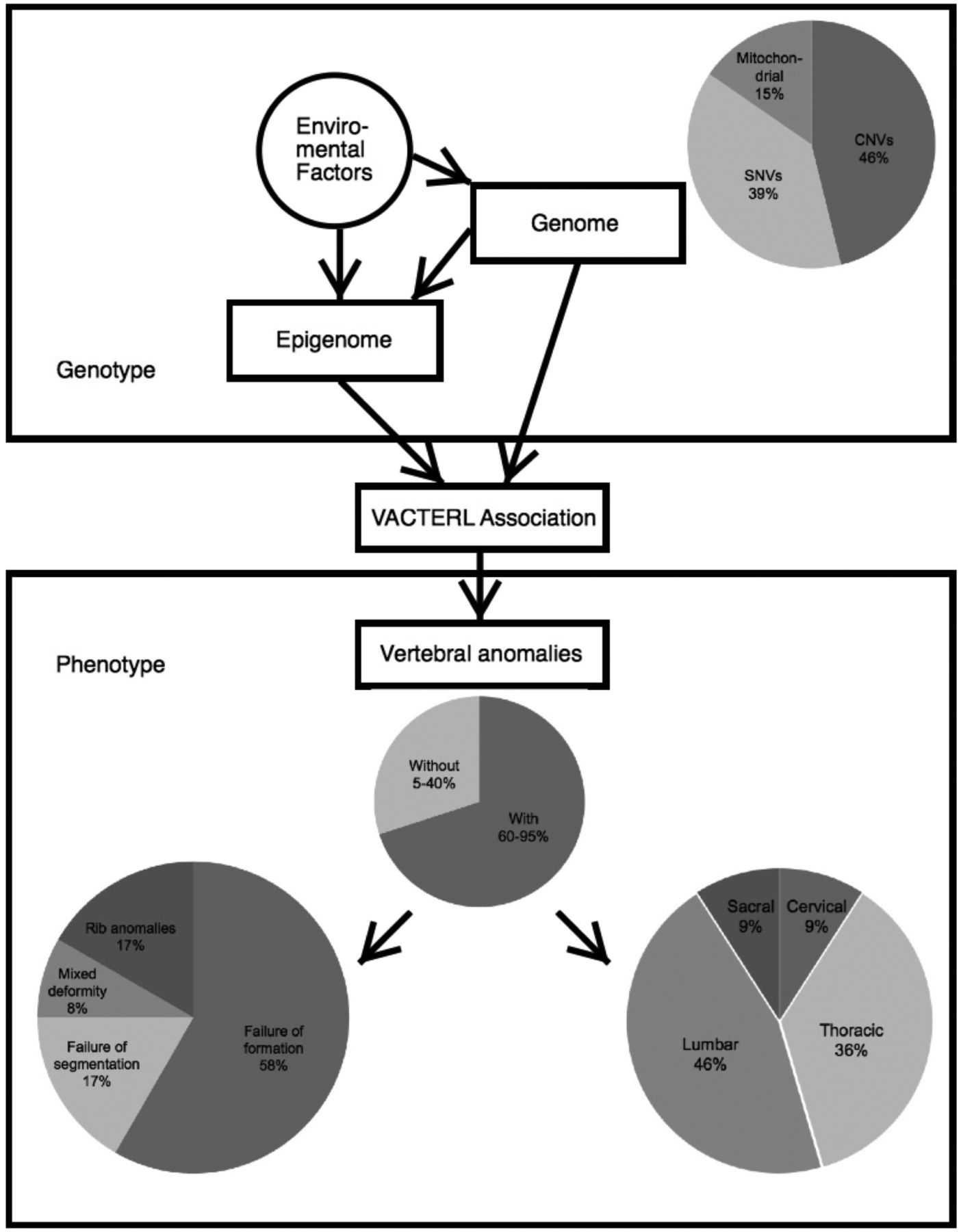
The aetiology of VACTERL association is not well understood (figure 2). As its phenotypes are too heterogeneous to be defined as a syndrome, and there is no major gene for this condition, thus it is still referred to as an ‘association’. The familial clustering phenomenon suggests a genetic role in its causality.34 ,41 ,42
{kind=link}
{kind=link}
General view of genetic findings and vertebral manifestations in VACTERL association. Mitochondrial, mitochondrial dysfunction; SNVs, single-nucleotide variants; VACTERL, vertebral anomalies (V), anal atresia (A), cardiac malformations (C), tracheo-oesophageal fistula (TE), renal dysplasia (R) and limb abnormalities (L).
X-linked VACTERL association by ZIC3 mutation
So far, the ZIC3 gene has been demonstrated to cause X-linked VACTERL association. Different types of ZIC3 mutations, including point mutations, deletions and polyalanine expansion, have been reported to be responsible for both VACTERL or VACTERL-like association.43–45 Cardiac defects are most commonly found as ZIC3 has important function in cardiac development and mutations in ZIC3 also cause X-linked heterotaxy (MIM#306955);43 ,46 ,47 anal atresia is present in most patients with ZIC3 mutations; vertebral anomalies are not commonly observed and demonstrated phenotypic variability.45 In animal models, Zic3 knockout mice mimic the human heterotaxy and cardiac phenotype with occasional vertebral/rib anomalies. Zic3expression was present at all stages of embryonic development within the anterior pre-somitic mesoderm but not in the developing anal region. Thus, anal atresia was not reported in Zic3-deficient mice,45 which differs from humans where anal atresia is also prevalent with ZIC3 mutations.
Sonic hedgehog pathway in VACTERL association
SHH gene has been implicated as the key inductive signal in patterning of the ventral neural tube, the anterior–posterior limb axis and the ventral somites.48 Studies on animal models indicate that sonic hedgehog (Shh) pathway is important for VACTERL association. Kim et al49 ,50 identified the first animal model that recapitulated the human VACTERL syndrome by knocking out genes (Shh and Gli) in Shh pathway. With different genes of the Shh signalling pathway affected, the mutant mice display various combinations, ranges and severity of the VACTERL phenotypes, implying a dosage-dependent effect. Furthermore, a VACTERL-like phenotype was reported in murine with a novel hypomorphic mutation in the Intraflagellar Transport Protein 172 (Ift172) gene.51 The Ift172gene encodes a component of the intraflagellar transport, which appears to play an active role in Shh signalling, and Ift proteins are required for both Gli activator and Gli repressor function.52 ,53
To the best of our knowledge, SHH or GLI3 mutations have not been identified in VACTERL patients.54 In humans, SHH mutation may cause more severe VACTERL phenotypes. Nowaczyk et al55 reported a patient with holoprosencephaly 3 and SHH haploinsufficiency who suffered from sacral anomalies (cleft S1, hemivertebra at S2 and absence of the rest of the sacrum and coccyx), genitourinary abnormality, multiple segments of bowel atresia and limb anomalies. Although this patient has a distinctive diagnosis, the phenotypic features overlap with VACTERL association. There is a possibility that SHH mutation causes these overlapping phenotypes.
Some genes that play roles in Shh pathway have been reported to be associated with VACTERL association. A heterozygous de novo 21bp deletion (c.163_183del) in the exon 1 of the HOXD13 gene,56 a downstream target of SHH,57 was identified in a 17-year-old girl, who was diagnosed with VACTERL association without vertebral anomalies. Another patient with rib anomalies diagnosed with VACTERL association was found with a 451 kb deletion at chromosome 3q28, which contains a single LPP gene.39 This gene encodes LIM domain containing preferred translocation partner in lipoma that has been shown to bind PEA3, an ETS domain transcription factor that has a role in regulating the SHH pathway.58 Moreover, CNV (microdeletions) as well as point mutation in FOXF1 gene have been identified in patients with VACTERL phenotypes.45 ,59 In animal models, Foxf1 has been found to be downregulated in Shh−/− mice60 ,61 and the Foxf1heterozygotes have been shown to display tracheo-oesophageal atresia and fistulas.62 ,63 Although HOXD13, LPP and FOXF1 mutation were sporadic findings in individuals,64 ,65 these studies argue in favour of that SHH pathway dysfunction is associated with VACTERL association.
Candidate gene mutations and CNVs
Several candidate gene mutations and CNVs have been reported to be related to VACTERL association (summarised in table 2). So far, these candidate gene mutations and CNVs listed are found mostly in sporadic cases, which need further large sample verification or functional experiments to confirm their pathogenicity.
Candidate genes and CNVs in VACTERL association
Although the genetic aetiology of VACTERL association has been far from established, previous studies did reveal some genetic mutations that can account for one or a few of the six CFs (table 2). For example, DLL3 gene, which encodes a ligand for the Notch signalling pathway that coordinates somitogenesis,66 has been found to cause block vertebrae in a Caucasian male VACTERL patient.67 Saisawat et al68 identified recessive mutations in the TNF receptor-associated protein 1 (TRAP1) gene in three families with VACTERL association. They also proved that Trap1 gene is highly expressed in the renal epithelia of 13.5-day-old mouse embryos and its mutations contribute to renal dysplasia.
Intriguingly, mutations of the same gene may cause variable expressivity among VACTERL patients, even within the same family. Dworschak et al69 identified chromosome 13q deletions in two patients with VACTERL phenotypes. The girl was born with perineal fistula, renal hypoplasia, bilateral triphalangeal thumbs and oligodactyly, butterfly vertebrae and cerebral anomalies, and died at 10 months of age. The second patient, a male child, suffered from perineal fistula, block vertebrae at C2–C3 and C4–C5–C6 and bilateral hearing loss. Pcsk5 gene has been identified as a candidate gene of VACTERL association in mice.70 Nakamura et al71 reported a Japanese VACTERL boy with eighth thoracic hemivertebra having a frameshift mutation of PCSK5, while his healthy father also shared the same mutation. Peddibhotla et al72 reported eight patients with chromosome 19p13.3 microdeletions and six of them fulfilled the diagnostic criteria for VACTERL association. Among the six VACTERL patients, one patient has vertebral anomalies while her two children, although with VACTERL association, are free from vertebral anomalies. These phenomena imply other modification factors desperate for further investigation in this condition.
Chromosomal aberrations
Chromosomal aberrations also contribute to VACTERL associations. Several case reports have been published that describe chromosomal anomalies in VACTERL patients as Felix et al77 and Brosenset al81 reviewed previously. However, chromosomal aberrations are not included here as they also contribute to the occurrence of congenital malformations beyond what is typically observed in VACTERL association.
Mitochondrial dysfunction
Damian et al82 first reported an A to G transversion in the mitochondrial NP3243 mutation in cystic kidney of a VACTERL child. Spinal radiograph showed multiple cervical and thoracic vertebral wedging, fusion and fission. She also had limb abnormalities, cardiac malformations and renal anomalies. This child belonged to a family in which other members had mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome and chronic progressive external ophthalmoplegia, which suggests mitochondrial dysfunction may contribute to VACTERL syndrome.83 Stone et al84 studied a cohort of 62 patients with VACTERL association and none of the affected children had measurable levels of the NP 3243 mutation. A few authors have previously reported an association of VACTERL association in patients with mitochondrial disorders known as complex IV respiratory chain deficiency.85–87 Overall, four of the five individuals presented with vertebral anomalies; three showed oesophageal involvement; two had anal atresia and two patients presented with additional minor dysmorphic features. Different combinations of other multiple congenital malformations have also been reported in a series of children with respiratory chain deficiency, leading to the hypothesis that in these patients congenital anomalies might result from an abnormal development during embryogenesis through either a lack of ATP or an alteration of apoptosis controlled by the mitochondrial machinery. However, it is also possible that mitochondrial dysfunction and congenital malformations in the patient described here are both secondary to an as yet unidentified process.88 In conclusion, whether mutation of mitochondrial dysfunction causes VACTERL association is still controversial. Some clinical signs and symptoms that may be not common in patients with VACTERL association, including progressive muscle weakness, characteristic patterns of cardiac, neurological and exocrine dysfunction,89 may suggest a potential existence of mitochondrial dysfunction.
In summary, the aetiology of VACTERL association appears to be heterogeneous, suggesting that it may be a complex condition. Besides the gene mutations and CNVs mentioned above, some other factors such as intronic mutations or epigenetic factors may also play important roles in this condition. Environmental factors including maternal diabetes90 and exposure to statins,91 which may associated with congenital anomalies, may play a significant role in the pathogenesis of VACTERL syndrome.
Conclusion
VACTERL association is a rare and complex condition with highly heterogeneous aetiology and manifestations. At the present time, there appears to be evidence for genetic factors contributing to VACTERL syndrome including single-gene mutations, CNVs and structure variants to mitochondrial dysfunction. Future studies are needed to identify epigenetics and environmental causes for VACTERL syndrome. Targeted genetic testing can contribute to eliminating overlapping diagnoses from further consideration in an affected individual. Notably, a given variant may explain a particular CF of VACTERL association, so it may be worth trying to investigate this sophisticated association by focusing on one of the six component features. ‘Vertebral anomalies’ is one of the core component features of VACTERL association, including formation and segmentation vertebral. Wu et al92 recently described a compound heterozygous model in which a null allele mutation in combination with a common haplotype of TBX6 causes congenital scoliosis, suggesting that genetic factors play an important role in vertebral anomalies. Additionally, we suggest that the genetic mutations may contribute to vertebral anomalies in a certain syndrome. Alternatively, VACTERL association may be caused by a ‘two-hit’ model in which two genes or one gene in combination with an epigenetic factor may elicit all associated features.93 In the future, combination of new genomic technologies such as whole-exome sequencing, whole-genome sequencing, comparative genomic hybridisation array and whole-genome bisulfite sequencing may well reveal a surprising number of additional contributing loci, delineating the entire spectrum of the VACTERL association in humans.
Acknowledgments
The authors thank Dr Pengfei Liu from the Department of Molecular and Human Genetics, Baylor College of Medicine, and Dr Xiaoyue Wang from the State Key Laboratory of Medical Molecular Biology, Chinese Academy of Medical Sciences, for their comments on the manuscript. They also express gratitude to the patient described in this article for his willingness to take part in this study.
References
Footnotes
Contributors YC, ZL and JC contributed equally to this article. YC, NW and ZW conceived and designed the review. YC interpreted data and contributed to the manuscript preparation. ZL and JC drafted the main manuscript. NW,ZW, YZ,GQ and PFG critically revised the manuscript and made comments on the structure, details and grammar for the article. SL, WC and GL contributed to data acquisition. All authors approved the final manuscript.
Funding The research was supported by National Natural Sciences Foundation of China (81501852, 81472046, 81271942, 81130034, 81472045), Distinguished Young Scholars of Peking Union Medical College Hospital (JQ201506), Beijing nova program (2016) and The Central Level Public Interest Program for Science Research Institue (No 13, 2015).
Competing interests None declared.
Ethics approval Ethics Committee of Peking Union Medical College Hospital.
Provenance and peer review Commissioned; externally peer reviewed.