Article Text

Abstract
Background Mucopolysaccharidosis VII (MPS VII) is an ultra-rare disease characterised by the deficiency of β-glucuronidase (GUS). Patients’ phenotypes vary from severe forms with hydrops fetalis, skeletal dysplasia and mental retardation to milder forms with fewer manifestations and mild skeletal abnormalities. Accurate assessments on the frequency and clinical characteristics of the disease have been scarce. The aim of this study was to collect such data.
Methods We have conducted a survey of physicians to document the medical history of patients with MPS VII. The survey included anonymous information on patient demographics, family history, mode of diagnosis, age of onset, signs and symptoms, severity, management, clinical features and natural progression of the disease.
Results We collected information on 56 patients from 11 countries. Patients with MPS VII were classified based on their phenotype into three different groups: (1) neonatal non-immune hydrops fetalis (NIHF) (n=10), (2) Infantile or adolescent form with history of hydrops fetalis (n=13) and (3) Infantile or adolescent form without known hydrops fetalis (n=33). Thirteen patients with MPS VII who had the infantile form with history of hydrops fetalis and survived childhood, had a wide range of clinical manifestations from mild to severe. Five patients underwent bone marrow transplantation and one patient underwent enzyme replacement therapy with recombinant human GUS.
Conclusions MPS VII is a pan-ethnic inherited lysosomal storage disease with considerable phenotypical heterogeneity. Most patients have short stature, skeletal dysplasia, hepatosplenomegaly, hernias, cardiac involvement, pulmonary insufficiency and cognitive impairment. In these respects it resembles MPS I and MPS II. In MPS VII, however, one unique and distinguishing clinical feature is the unexpectedly high proportion of patients (41%) that had a history of NIHF. Presence of NIHF does not, by itself, predict the eventual severity of the clinical course, if the patient survives infancy.
- Clinical genetics
- Genetics
- Metabolic disorders
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Mucopolysaccharidosis VII (Sly syndrome; MPS VII) is an autosomal recessive lysosomal storage disorder (LSD) that is characterised by the deficiency of activity of β-glucuronidase (GUS: β-D-glucuronoside glucuronosohydrolase, Enzyme Commission (EC) number: 3.2.1.31; GUSB: MIM 611499).1 ,2 GUS is one of the enzymes involved in the stepwise degradation of three glycosaminoglycans (GAGs). In the absence of GUS, chondroitin sulfate, dermatan sulfate and heparan sulfate are only partially degraded and the partially degraded fragments accumulate in the lysosomes of many tissues, eventually leading to cellular and organ dysfunction.
MPS VII is an ultra-rare disorder and precise epidemiological data are scarce. In an epidemiological study of the MPS population in northern Ireland from 1948 to 1985, Nelson reported three cases of fatal non-immune hydrops fetalis (NIHF) in infants born to consanguineous parents that were believed to be MPS VII due to GUS deficiency on the basis of placental histology and enzyme assays of both parents. However no living cases of MPS VII were reported.3 In the 1970s a total of 10 cases of MPS VII were reported in the literature.1 ,4–9 Subsequently, Meikle reported two patients born with MPS VII from 1980 to 1996 in Australia10 and Applegarth reported three cases in British Columbia from 1969 to 1996.10 ,11 In The Netherlands, Poorthuis reported six cases of patients born with MPS VII from 1985 to 1995.12 No cases were reported in Germany (1980–1995), Scandinavian countries (1975 and 2004), Taiwan (1984–2004) and western Australia (1969–1996).13–16 Recently one more case with hydrops fetalis was reported in Germany.17 Overall the frequency of this disease is estimated to be 1:300 000–1:2 000 000.18 Many patients may have been missed because of early death in utero since the most frequent phenotype may be hydrops fetalis in the antenatal form.12 ,17 ,19–21 Others may have died in early infancy without diagnosis.
The GUS gene is located on chromosome 7q11.21–7q11.22 spanning 20 kb and containing 12 exons.22 Isolation and characterisation of the human GUS cDNA facilitated the investigation of allelic heterogeneity in patients with MPS VII.23–27 To date 49 unique, disease-causing mutations have been reported in patients with MPS VII. Mutations are distributed along the whole gene and include missense mutations (78.6%), nonsense mutations (12.6%), deletions (5.8%) and splice-site mutations (2.9%).28–32 In addition to reports of disease-causing mutations, there are two reports of a pseudo-GUS deficiency where obligate heterozygotes have unusually diminished enzyme activity, and increased excretion of urinary GAGs without pathological manifestations.33 ,34 Clinical severity observed in patients with MPS VII and the correlation with their genotype currently remains an important area of investigation.
The clinical presentation and disease progression of MPS VII span a wide severity spectrum. Most patients with MPS VII have cognitive impairment, hepatosplenomegaly and skeletal dysplasia. However, affected patients show a wide range of clinical variability, from early, severe, multisystem manifestations to a milder phenotype with later onset and normal or near-normal intelligence. Although patients with MPS VII may present with hydrops fetalis at birth and only survive a few months, rare patients with milder manifestations of MPS VII have survived into the fifth decade of life.2 ,4
As with other MPS diseases, as more patients were identified, it became clear that there is a broad spectrum or continuum of disease severity among patients with MPS VII. To date, accurate assessments of the frequency and clinical characteristics of the disease have been difficult. Gathering data on the natural history of MPS VII was needed to understand more fully the spectrum of disease severity, and the rate of progression and distribution of specific symptoms in untreated patients with MPS VII. In this study, data were collected on disease severity, the rate of progression and distribution of specific symptoms in untreated patients with MPS VII. These data are important to enable early detection, to define potential clinical end points and to provide comparative data to judge the effects of future treatments.
Subjects and methods
Survey and study population
A survey was developed to document the medical history of patients with MPS VII. The survey included anonymous information on patient demographics, family history, mode of diagnosis, age of onset, signs and symptoms, severity, management, clinical features and natural progression of the disease. The survey was administered to physicians who currently or previously cared for patients with MPS VII. The database of physicians was provided by Engage Health and an email was designed to recruit physicians from all over the world. All physicians who participated were contacted by phone and each interview lasted approximately 1 h. Since the information obtained was anonymous and no protected health information was collected, Institutional Review Board (IRB) approval was not obtained. Information was collected on 56 patients from 11 countries. Data were collected from 23 physicians from North and South America, Europe, Asia and Oceania. The survey was conducted in English and/or Spanish. On each item, physicians responded: yes, no, or not reported in patient's record. From the items with informative responses (yes/no) we calculated the percentage for each answer.
Alignment of sequences
Multiple amino acid alignment of GUS sequences from human, chimpanzee, cow, pig, dog, mouse, rat, chicken, frog, fruit fly, mosquito, honey bee, red flour beetle, Gram-positive bacteria and enterobacteria was constructed by using ClustalW Multiple alignment application on BioEdit program35 with manual adjustment. The final selected alignment was converted to Molecular Evolutionary Genetics Analysis (MEGA) 4.36 GenBank reference sequences: Homo sapiens, NM_000181.3; Pan troglodytes, XM_001138883.4; Bos taurus, NM_001083436; Sus scrofa, AK232674; Canis lupus familiaris, NP_001003191.1; Mus musculus, NP_034498; Rattus norvegicus, NP_058711; Gallus gallus, NP_001034405.1; Xenopus tropicalis, CT030620; Drosophila melanogaster, NP_001014535.1; Anopheles gambiae, XP_320660.4; Apis mellifera, XM_393305; Tribolium castaneum, XM_964260; Arthrobacter Sp. RP10, AAV91790; and Escherichia coli, AAB30197.
Data handling and analysis
After the information was collected, all surveys were compiled in a database for statistical analyses. Although all the information from physicians was compiled, information on some items could not be obtained. Variables were summarised using descriptive statistics including mean, median, ranges, percentages and/or frequencies. All analyses were performed using SPSS for windows (IBM SPSS Statistics for Windows, V.20.0. Armonk, New York: IBM Corp).
Results and discussion
Patient demographics
Our survey collected data on 56 patients of whom 53% were boys, 36% girls and 11% sex-unknown (diagnosed in utero). Sixty-two per cent of the patients were Caucasian. The other patients were of white-Hispanic (18%), black (9%), Asian (7%) and other (4%) origin. The geographical distribution of patients with MPS VII (table 1) showed they were predominantly from Brazil (27%), USA (20%), Germany (18%) and Argentina (11%). The high percentage of patients from Latin America may reflect ascertainment bias because of national practices of referring all patients with lysosomal storage diseases to key treatment centres.
Demographics of patients with MPS VII (n=56)
Information on patient age at the last visit to the physician was collected in 28 patients (table 1). The mean age distribution at the last visit to the physician was 13.25 years, the median was 11 years and the distribution of the ages was wide. Fifty-three per cent of the patients were still living at the time of the physician interview.
Diagnosis and presentation of the disease
Thirty-four per cent of the patients were diagnosed prenatally or below 1 year of age, 16% from 1 year to 3 years of age, 11% from 3 years to 5 years of age, 20% from 5 years to 10 years of age and 12% over 10 years of age. Age at time of initial diagnosis in 7% of the patients was unknown.
Patients with MPS VII were classified based on their phenotype into three different groups: (1) non-immune neonatal hydrops fetalis (NIHF) (n=10), (2) Infantile or adolescent form with history of hydrops fetalis (n=13) and (3) Infantile or adolescent form without known hydrops fetalis (n=33). Thirteen patients with MPS VII who had the infantile form with history of hydrops fetalis and survived childhood, had a range of clinical manifestations from mild to severe, suggesting that presence of hydrops fetalis does not necessarily predict the subsequent severity of the disease (figure 1).

Serial pictures of a male patient who survived a stormy early course, beginning with neonatal hydrops and subsequently progressed more slowly with cognitive impairment, hepatosplenomegaly, obstructive airway disease, heart valve abnormalities and dysostosis multiplex including progressive hip dysplasia. This patient died undergoing anaesthesia from a dental procedure at age 12 years, which is a complication from which these patients often suffer. (A) 2.5 months old, (B) 6 months old, (C) 1 year old, (D) 3 years old, (E) 5 years old, (F) 6 years old, (G) 7 years old, (H) 8 years old, (I) 9 years old and (J) 11 years old. Photographs of the deceased patient were obtained and approved for publication with maternal consent. X-rays are shown below in the section clinical course of the disease (figures 12 and 13).
Clinical characteristics of patients with infantile or adolescent form of the disease
Distribution of clinical symptoms was tabulated for 46 patients with MPS VII, of whom 13 had a history of hydrops. From the items with informative responses (yes/no), we calculated the percentage, and the number of informative responses varies for each item as noted in each figure.
Head, eyes, ear-nose-throat (ENT) (figure 2): Coarse facial features were reported in 87% of patients, 87% had increased cranial circumference and 78% had a short neck (figure 2A). Coarse hair was noted in 60% of the patients.
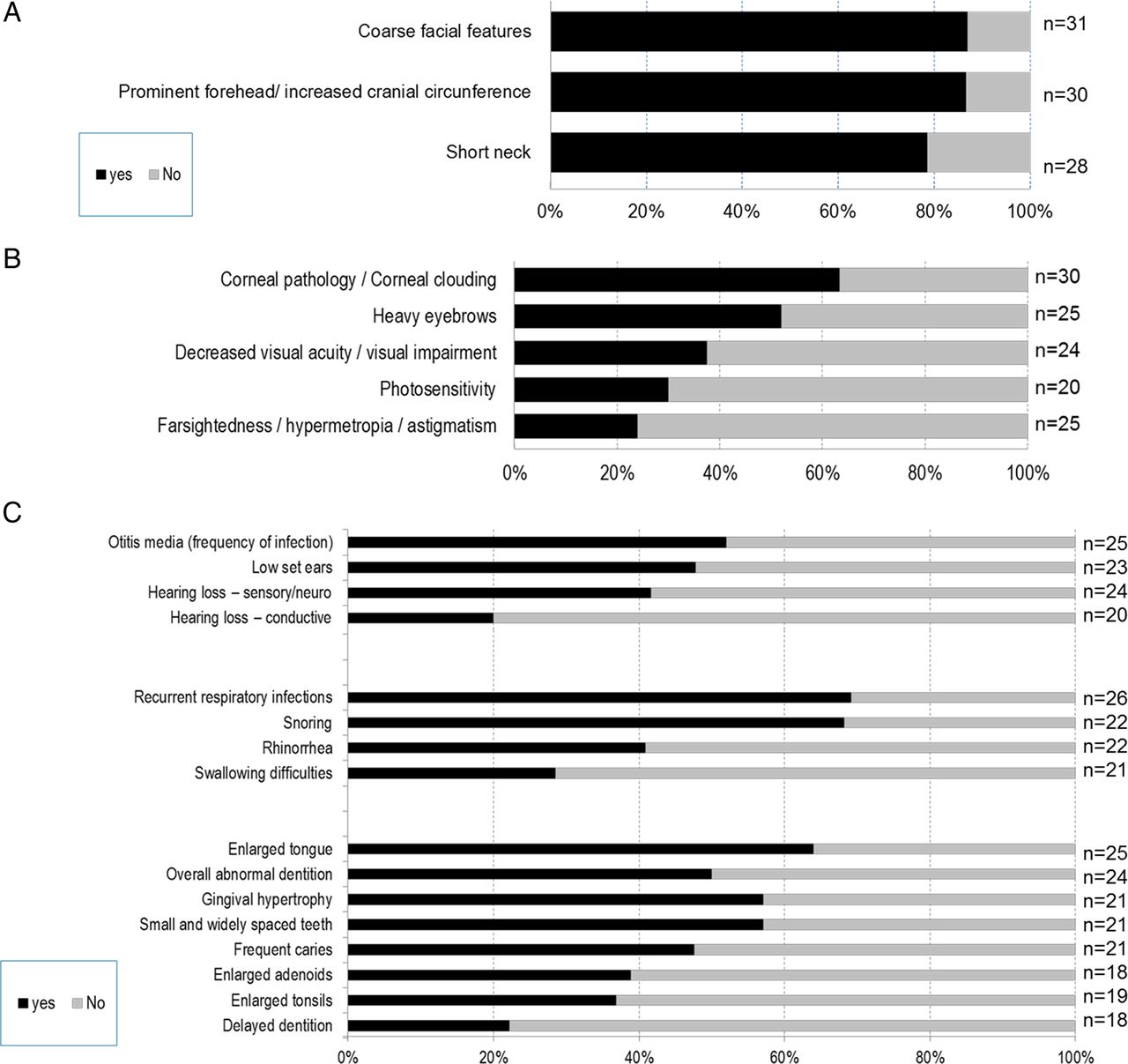
Clinical symptoms found in (A) head and neck, (B) eyes, and (C) ear, nose and throat of patients with mucopolysaccharidosis VII (MPS VII) disease.
The predominant ocular features were corneal clouding (63%), heavy eyebrows (52%), visual impairment (37%) and photosensitivity (30%) (figure 2B). A majority of patients had ear (52%) and respiratory (69%) infections, enlarged tongue (64%), which is associated with snoring (68%), abnormal dentition (50%) with small and widely spaced teeth (57%), and gingival hypertrophy (57%). Sensorineural hearing loss was observed in 41% of the patients (figure 2C).
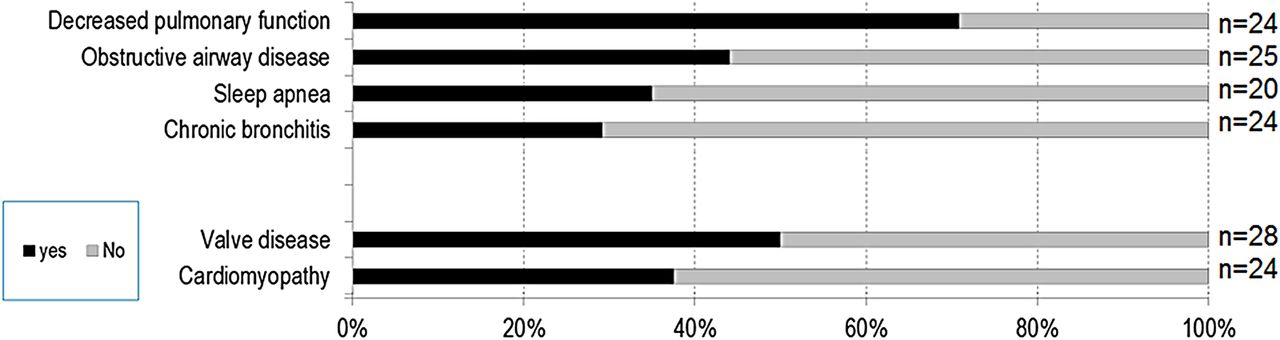
Lungs and heart (figure 3): Clinical symptoms related to the respiratory system included decreased pulmonary function (71%), obstructive airway disease (44%), sleep apnoea (35%) and chronic bronchitis (29%). Heart abnormalities included cardiac valve disease (50%) and cardiomyopathies (37%).

Clinical symptoms related to lungs and heart.
Musculoskeletal (figure 4): Dysostosis multiplex on X-ray (90%) was the most consistent finding in the MPS VII patient survey. Next was loss of joint range of motion (85%), giving rise to restricted mobility (78%), joint contractures (84%) and stiffness (72%). Spine deformities included scoliosis (69%), kyphosis (68%) and gibbus (63%). Leg deformities included genu valgum (63%) and talipes equinovarus (35%). Hand abnormalities included a decreased range of wrist motion (58%), clawed hands (56%) and curved fingers (54%). Acetabular dysplasia in hips was observed in 53% of the patients and 63% had hip or back pain when bending over.
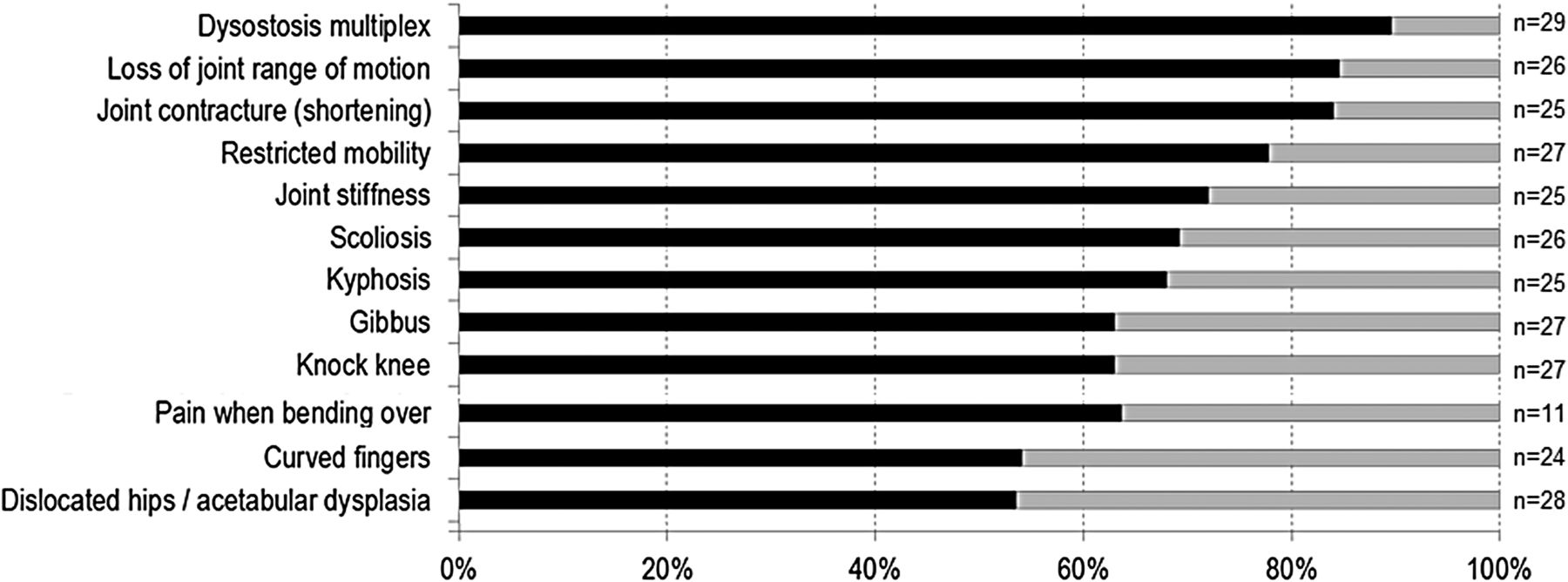
Musculoskeletal signs and symptoms in patients with mucopolysaccharidosis VII (MPS VII).
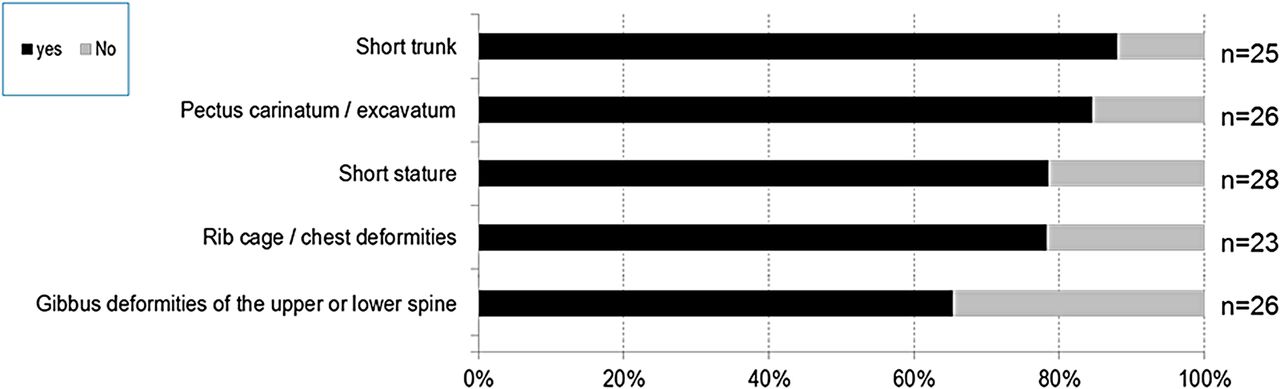
Thoracolumbar and abdominal abnormalities (figure 5): 88% of patients had a short trunk, 85% had pectus carinatum or excavatum, 78% had rib cage/chest deformities and 79% had short stature. Additionally, 75% of patients had hepatomegaly/splenomegaly and 61% had umbilical and/or inguinal hernias.

Thoracolumbar abnormalities in patients with mucopolysaccharidosis VII (MPS VII).
Neurological (figure 6): Limited vocabulary (94%) and mental retardation (86%) were the most common neurological problems in patients with MPS VII.

Major neurological features of patients with mucopolysaccharidosis VII (MPS VII).
Growth
Height: Short stature was one of the most characteristic phenotypes observed in patients with MPS VII. In this survey, the preliminary height measurements indicate that patients have normal growth until 18–24 months of age (figure 7A).
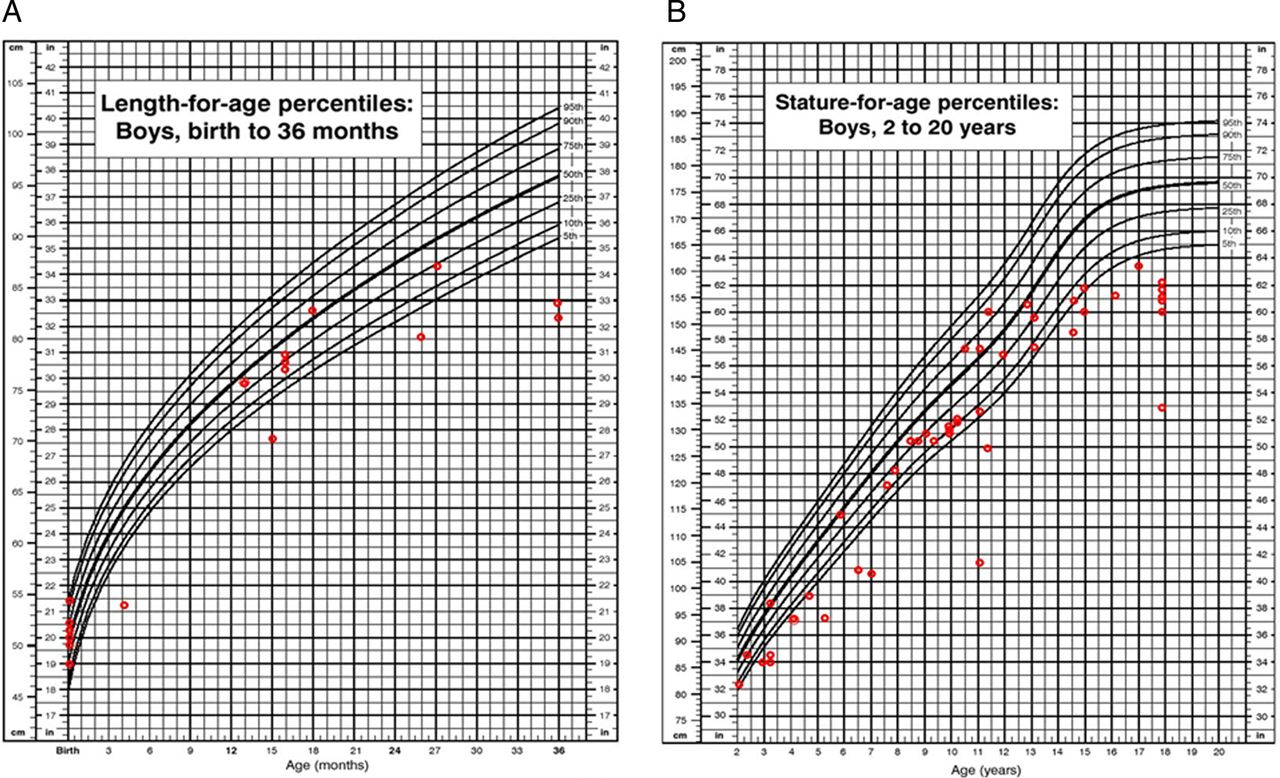
(A) Length for age in boys (0–36 months) affected with mucopolysaccharidosis VII (MPS VII). (B) Stature for age in boys (2–18 years) affected with MPS VII. Data are compared with normal growth charts from the CDC.
In boys we observed scattered values of height (between 5th and 90th centiles) from 2 years of age to 13 years. Thereafter, we observed a rapid decline in growth (ie, height values compared with normal controls at or below the 5th centile). In boys at 18 years old, short stature was pronounced. We found a mean height of 151.8±7.9 cm (n=7), corresponding to a Z score of −3.3 when compared with age-matched Centers for Disease Control and Prevention (CDC) controls (figure 7B).
In girls the short stature was even more extreme. We observed a clear decrease in attained height values when compared with normal controls after 24 months of age with values below the 25th centile. At 18 years of age the mean height was 114.3±25.4 cm (n=5), corresponding to a Z score of −7.8 when compared with age-matched CDC controls (figure 8).

(A) Length for age in girls (0–36 months) affected with mucopolysaccharidosis VII (MPS VII). (B) Stature for age in girls (2–18 years) affected with MPS VII. Data are compared with normal growth charts from the CDC.
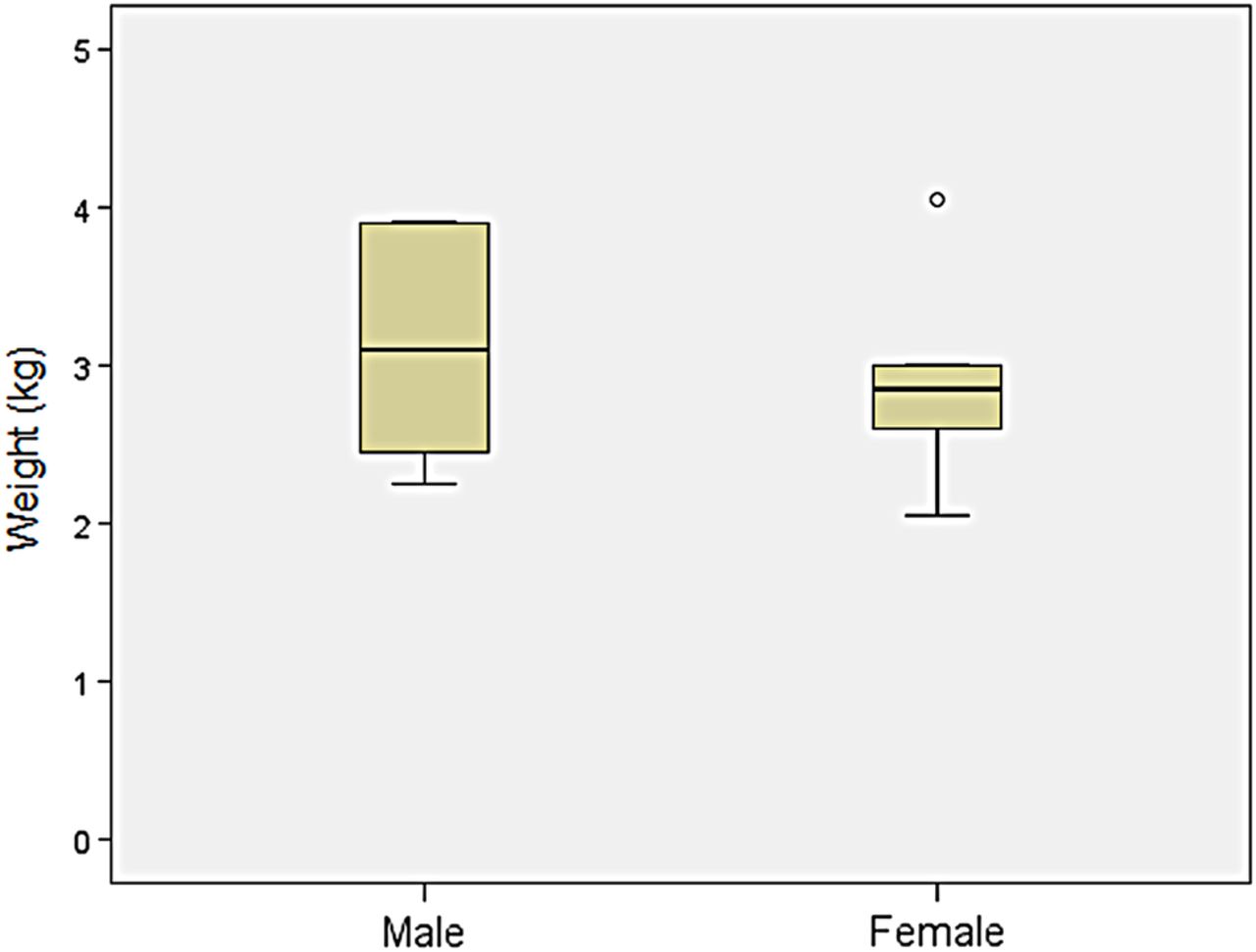
Weight: Comparison of birth weight between patients with MPS VII and age-matched controls found the mean birth weight for boys with MPS VII to be similar (3.4±0.6 kg) (n=18) compared with the CDC control charts for boys (3.5±0.5 kg). The mean birth weight for girls with MPS VII was 2.9±0.7 kg (n=5), which was slightly lower than the CDC values for girls (3.4±0.4 kg) (Z score=−1.0), the caveat being that the sample size was small (figure 9).
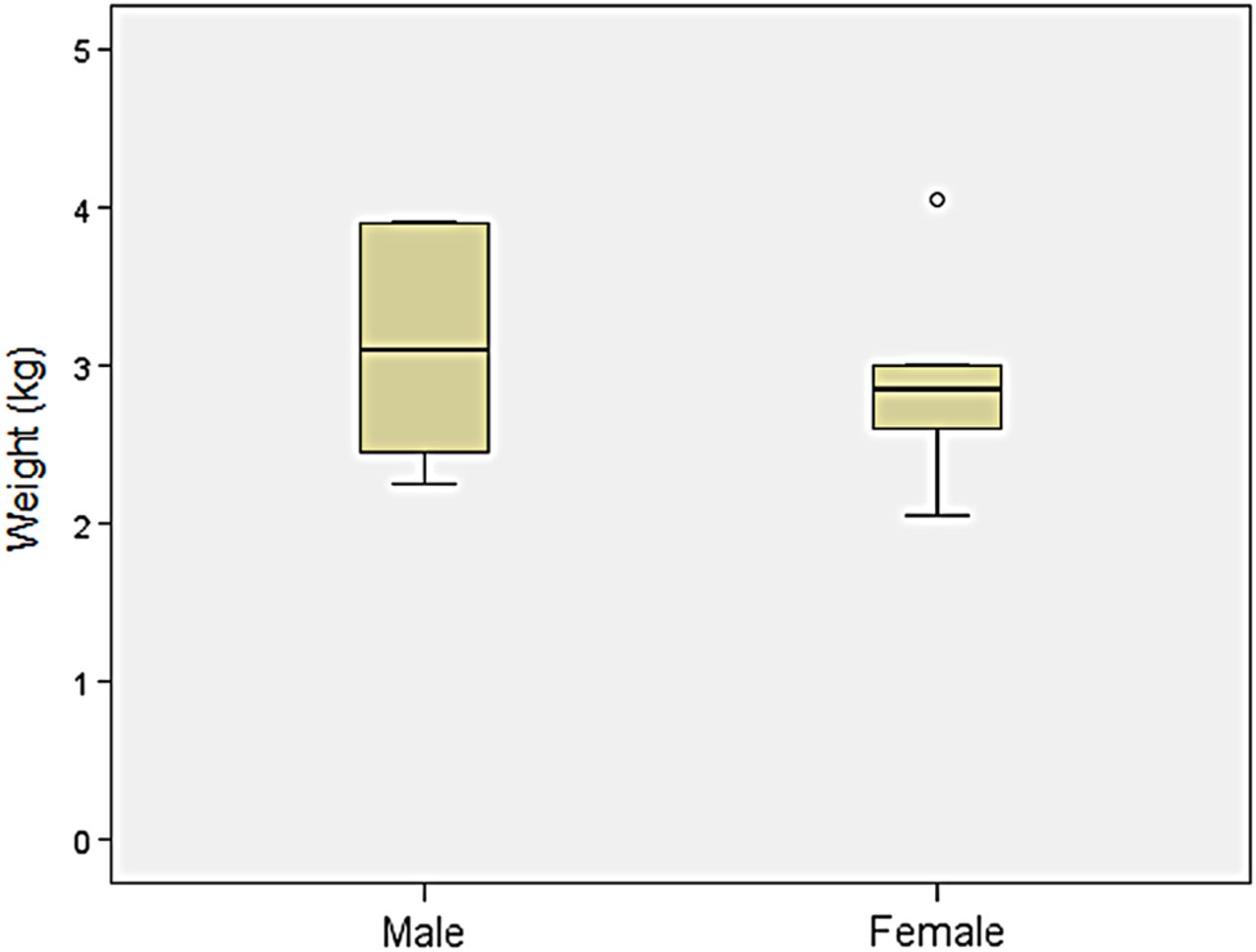
Comparison of birth weight of patients with mucopolysaccharidosis VII (MPS VII).
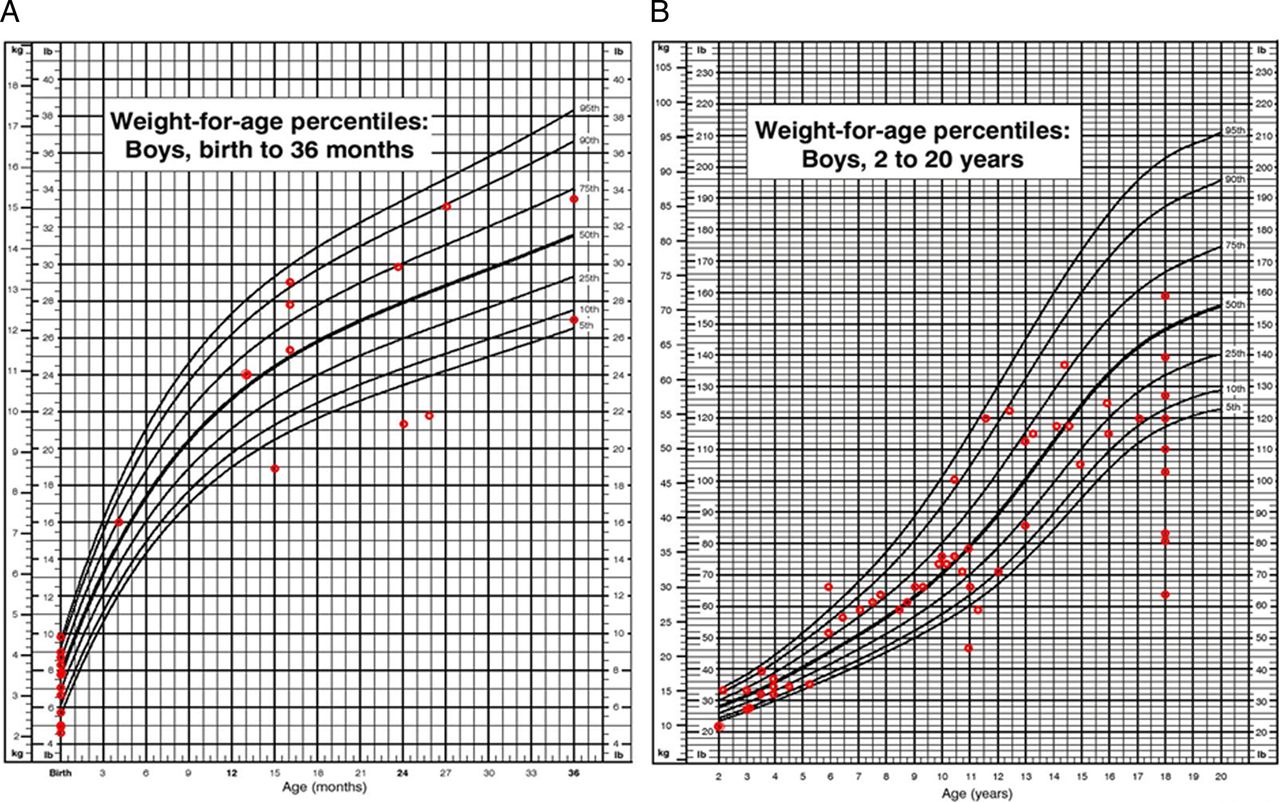
At 18 years of age, the mean weight of boys with MPS VII was 50.1±13.4 kg (n=10), which corresponds to a Z score of −2.1 (2nd centile) (figure 10). The average body mass index (BMI) for boys with MPS VII at ≥18 years was not abnormal: 23.5±4.8 kg/m2 (n=13). One male patient (21 years old) had a very high BMI of 30.2 kg/m2, which indicated obesity (96.6 centile). Four patients of different ages (2 years, 4 years, 6 years and 11 years old) had underweight BMIs (figure 10).

(A) Weight for age in boys (0–36 months) affected with mucopolysaccharidosis VII (MPS VII). (B) Weight for age in boys (2–18 years) affected with MPS VII. Data are compared with normal growth charts from the CDC.
In comparison, the mean weight of girls with MPS VII at 18 years of age was 36.2±12.0 kg (n=4), which corresponds to a Z score of −4.2 (figure 11). The average BMI for girls over 18 years with MPS VII was 23.7±0.2 kg/m2 (n=8); only one female patient (11 years old) had a high BMI of 25.3 kg/m2 which indicated obesity (97.3 centile). Three patients of different ages (4 years, 6 years and 15 years old) had underweight BMIs (figure 11).

(A) Weight for age in girls (0–36 months) affected with mucopolysaccharidosis VII (MPS VII). (B) Weight for age in girls (2–18 years) affected with MPS VII. Data are compared with normal growth charts from the CDC.
Genotype and phenotype
In this study only 10.7% of patients with MPS VII (n=6) had mutational analysis, which makes it difficult to describe an accurate correlation between genotype and phenotype (table 2). Three patients had mutations that were previously described (p.L176F, p.R382C, p.P408S and p.P415L) associated with an attenuated phenotype.29 The phenotype observed in these patients ranges from mild with mental retardation to severe with moderate mental retardation. One patient was compound heterozygote for the mutation p.D362N which has been associated with a severe phenotype.29 However, this patient underwent bone marrow transplantation (BMT, Patient 5), thus association between phenotype and genotype was not possible. Two other patients with severe phenotype were compound heterozygotes for mutations that have not been reported yet (p.D50Y, pV99M, p.W289X and p.L565V), hence detailed correlation between phenotype and genotype has not been defined. Most of the residues that were mutated had a high degree of conservation when compared with sequences of other species (table 2).
Mutations of patients with MPS VII in the survey (n=6)*
Clinical course of the disease
The first signs and symptoms observed in patients with MPS VII included hydrops fetalis, hernia (umbilical or inguinal), coarse face, hepatosplenomegaly, skeletal dysplasia, heart and respiratory problems, mental retardation, and ear infections. Of the 23 patients with hydrops fetalis in this study, 10 patients had prenatal-onset NIHF. Of those with prenatal-onset hydrops that survived gestation, most died in early infancy of heart, kidney or respiratory failure (table 3). Thirteen patients had the infantile or adolescent form of the disease with history of hydrops fetalis. The 12 patients with the infantile form of the disease who had recovered from hydrops manifested characteristic symptoms of the MPS VII disease, varying from mild to moderate or severe phenotype, as reported by the physicians. This indicates that the presence of neonatal hydrops fetalis does not, by itself, predict the eventual severity of the disease.
Patients with MPS VII from the survey with history of neonatal non-immune hydrops fetalis (NIHF)
Progression of the disease beyond infancy was somewhat variable. Patients with mild or moderate early manifestations tended to deteriorate more slowly than patients with more severe manifestations. The clinical course of the disease at the last visit with the physician was reported as ‘slowly worse’ in 78% of the patients, ‘stable’ in 15% and ‘rapidly worse or other’ in 7% of the patients (n=27).
In agreement with the literature, repeated upper respiratory and pulmonary infections which required antibiotics were common in the first 2 years of life, after which there was a decrease in the frequency of infection with age. Mild to moderate mental retardation was common and usually required special education. Hearing impairment was common and likely contributed to delayed speech development. Skeletal deformities of the spine and chest wall were common and progressed gradually with age (figure 12). Most patients had hepatomegaly with limited excursion of the diaphragm which possibly contributed to respiratory insufficiency. Typically, hip dysplasia was associated with pain on walking which led to wheelchair dependency by age 10 years in some patients (figure 13). Prominent cardiac murmurs were common but serious valvular insufficiency was not.
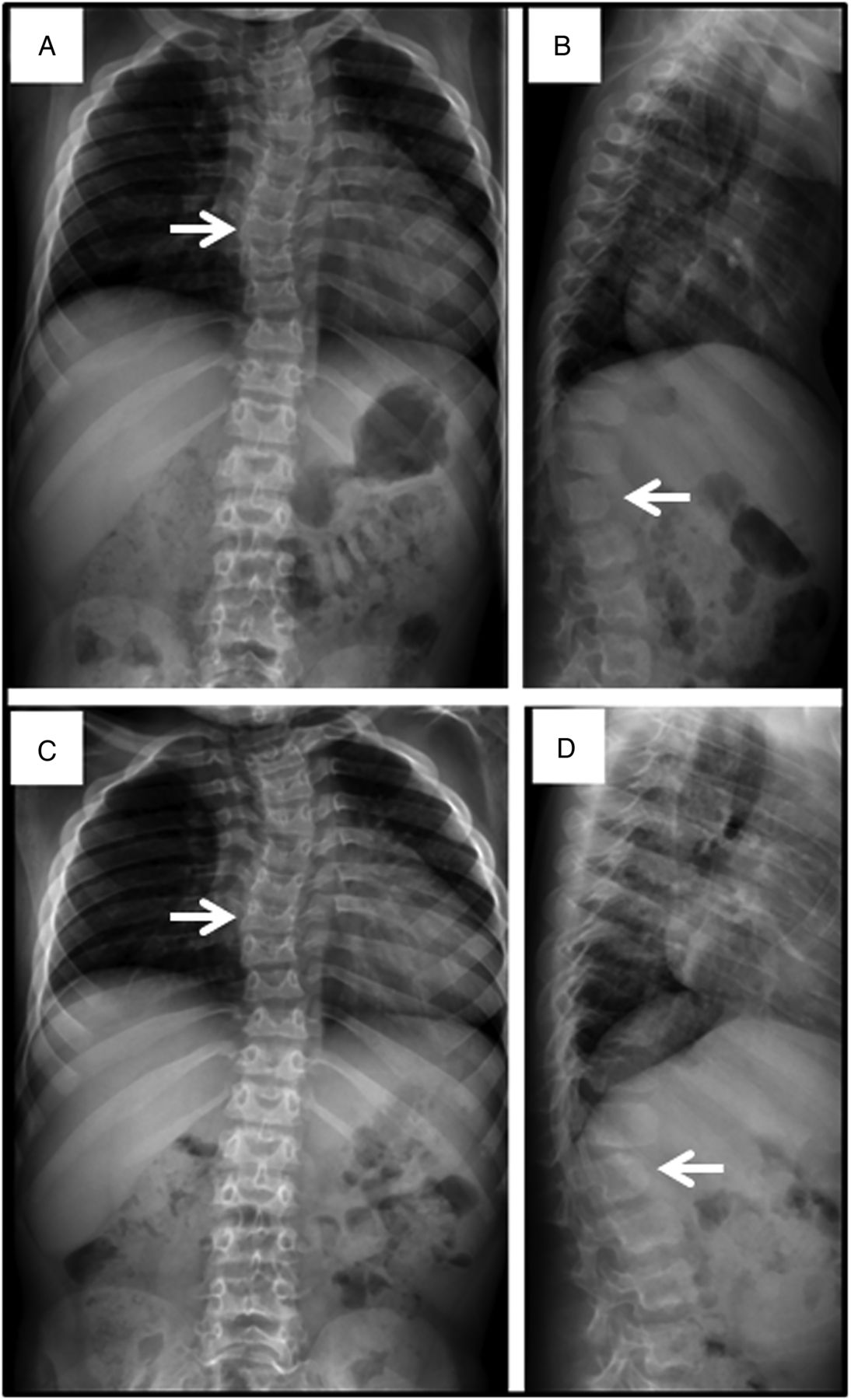
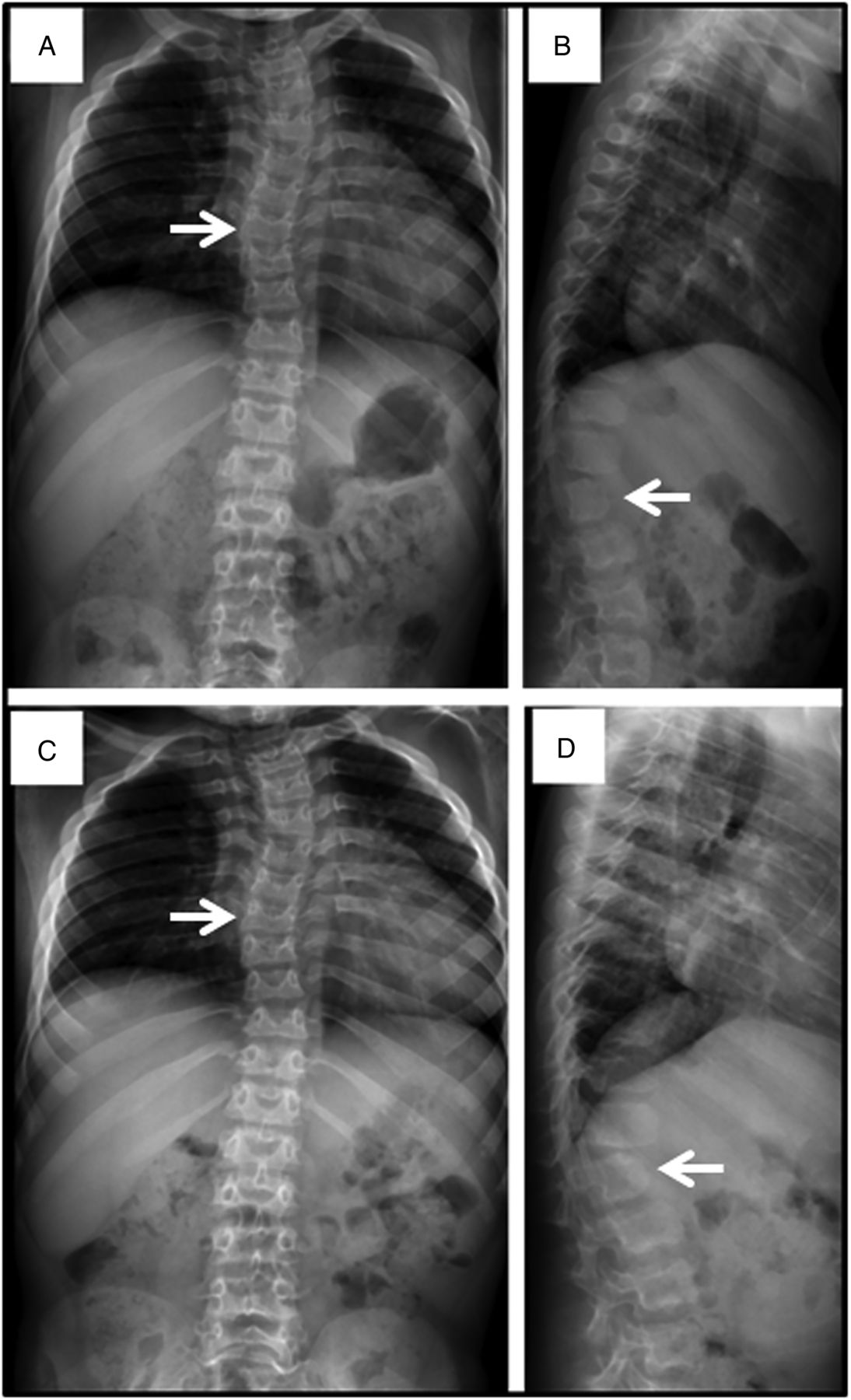
Spine films in a patient with mucopolysaccharidosis VII (MPS VII) demonstrate findings common to all forms of MPS at age 7 years (A and B) and age 8 years (C and D). Anteroposterior views (A and C) reveal mild scoliosis (arrow) and broad ribs. The lateral films (B and D) demonstrate irregular anterior vertebral body growth, particularly at the L2 level (arrow).
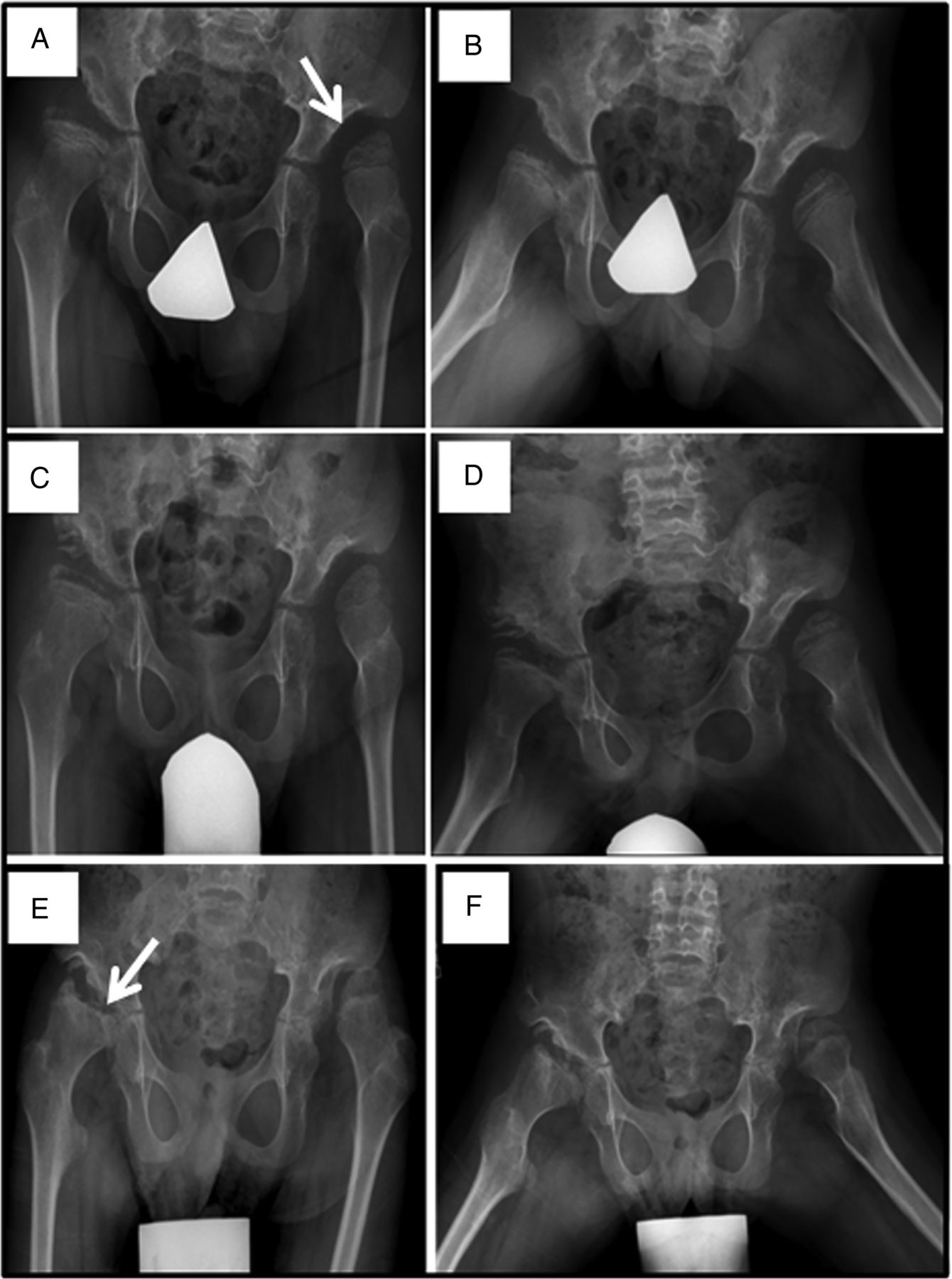
Progressive hip dysplasia is seen in this patient with mucopolysaccharidosis VII (MPS VII). At age 7 years (A), the right hip shows subluxation with poor coverage, while the left hip is already dislocated and sits in a pseudoacetabulum (arrow). By age 8 years (C), an acetabular shelf procedure has been performed and by age 10 years (E), has not prevented further subluxation and femoral head erosion (arrow).
Most patients underwent palliative, symptomatic-care treatments to improve their quality of life. These included surgeries, antibiotic treatment and physical therapy, among others. About 46% of the patients with the infantile or adolescent form of the disease had surgeries. The most common included: hernia repair, hip replacement and cervical fusion (figure 14). Seventeen per cent of the patients were able to perform daily activities by themselves, but more often, some assistance was required. Almost half of the patients with the infantile or adolescent form of the disease (46%) were able to walk by themselves without assistance. However, most patients lost this ability with time due to hip dysplasia. Common causes of death included complications of hydrops fetalis with respiratory and renal failure, heart disease, decreased pulmonary function and/or obstructive airway disease, complication of BMT and aspiration (figure 15).

Distribution of surgical operations performed on patients with mucopolysaccharidosis VII (MPS VII) (n=21, 45.6% of total number of patients).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Distribution of causes of death by age. Causes included: Prenatal to 1 year, (50%) complications of hydrops fetalis, respiratory failure and renal failure; ages 1–5 years (5%), obstructive airway disease; ages 5–10 years (15%), heart disease, complications of bone marrow transplantation (BMT) and pulmonary failure; ages 10–15 (5%), decreased pulmonary function; and over 15 years of age, heart disease, pulmonary failure and/or obstructive airway disease, and aspiration/heart attack.
Treatment
Bone marrow transplantation
In our survey we identified five patients that underwent BMT (table 4). Three survived.
Patients from the survey that underwent bone marrow transplantation (BMT)
BMT Patient 1, Spanish Caucasian girl, diagnosed at 1year 10 months of age. Onset of the disease was observed at birth due to the presence of talipes equinovarus and kyphosis. At 2 years of age BMT was performed, but failed with autologous reconstitution. At 4 years of age, BMT was performed once more with successful engraftment. At 4 years and 1 month of age, the patient had moderate clinical manifestations of the disease with normal intelligence and slow progression of the disease. The patient has coarse facial features, short neck, heavy eyebrows, low set ears, exophthalmos and skeletal abnormalities including short stature, short trunk, pectus carinatum, joint contractures, genu valgum, kyphosis, scoliosis and talipes equinovarus. The result of the BMT on subsequent course is unknown.
BMT Patient 2, Brazilian black boy, diagnosed at 2 years of age. He had severe manifestations of the disease including mental retardation, coarse facial features, prominent forehead, prognathism, corneal clouding, visual impairment, recurrent respiratory infections, macroglossia, cardiac valve disease and decreased pulmonary function. He underwent BMT after 7 years of age, but subsequently died from complications of the procedure.
BMT Patient 3, Brazilian black boy, diagnosed at 4 months of age because of history of an older affected sibling. Initial symptoms included hepatomegaly and splenomegaly. He had severe manifestations of the disease with mental retardation. He had coarse facial features, prominent forehead, prognathism, corneal clouding, visual impairment, macroglossia, cardiac valve disease, short stature, short trunk, gibbus and deformity of the thorax, recurrent respiratory infections, decreased pulmonary function, hepatosplenomegaly, umbilical hernia, joint contractures and stiffness, restricted mobility, loss of joint range of motion, clawed hands, language impairment and hyperactivity. The patient died a few years after the procedure.
BMT Patient 4, Spanish Caucasian boy had prenatal hydrops fetalis at 22 weeks of gestation but MPS VII was not diagnosed until 26 months of age. Initial signs and symptoms included difficulty breathing, hepatosplenomegaly, inguinal hernia, coarse facial features, prominent forehead, mental retardation, developmental delay and short neck. He underwent BMT at 3 years of age. At 15 years of age, he showed a moderate clinical phenotype suggesting the disease may have stabilised somewhat after BMT. However, at the last exam he showed coarse facial features, short neck, corneal clouding, visual impairment, photosensitivity and dry eyes. After BMT, he still had otitis media, swallowing difficulties and recurrent respiratory infections. He also had abnormal dentition with caries, thin enamel, gingival hypertrophy and macroglossia. Skeletal abnormalities included short stature, short trunk, gibbus, joint contractures, odontoid hypoplasia, scoliosis, kyphosis, genu valgum, curved fingers, clawed hands, restrictive mobility, loss of joint range of motion, decreased wrist range of motion and thorax deformities. This patient also had restrictive and obstructive airway disease, asthma, chronic bronchitis and decreased pulmonary function. The patient was hydrocephalic with language impairment. He also had atopic dermatitis.
BMT Patient 5, American Caucasian girl, had hydrops fetalis, fetal cardiac distress, tachycardia. MPS VII was diagnosed at 2 weeks of age and she underwent BMT at 7 months of age. At age 15 months she had no clinical manifestations. Hepatomegaly had disappeared. Intelligence appeared normal and she achieved normal developmental milestones, including walking at 1 year of age. The patient exhibits a prominent forehead, and some cardiomyopathy with mild atrial enlargement.
Enzyme replacement therapy
Enzyme replacement therapy (ERT) Patient 1, Asian-American boy, had hydrops fetalis and splenomegaly. He was diagnosed at 18 months of age when he presented with severe cord compression for which he underwent cervical fusion.37 The patient eventually learned how to walk and attended school but lost the ability to walk as the disease progressed. He suffered from upper airway obstruction, progressive heart valve disease and hepatosplenomegaly. The main complications involved his pulmonary status. He was hospitalised several times for respiratory failure and elevated end-tidal carbon dioxide levels of 80 mm Hg or more.38
Due to the patient's pulmonary failure despite usage of maximum resources to improve his pulmonary function, an emergency request for compassionate use of investigational recombinant human GUS (rhGUS) therapy was made and granted by the US Food and Drug Administration. The enzyme was provided by Ultragenyx Pharmaceutical.
The patient was treated with intravenous infusions of 2 mg/kg of rhGUS every other week. After 24 weeks of treatment, no serious adverse effects were noted. The levels of GAGs in the urine decreased approximately 60% from the baseline, spleen and liver size declined, and pulmonary function improved as evidenced by increased tolerance for off-ventilator challenges. Quality of life was improved and he regained the ability to attend school and to eat some food orally. This patient's experimental treatment with ERT is still ongoing. A report summarising the clinical course for ERT Patient 1 to date was recently published.38 Additional formal clinical trials of ERT on patients with MPS VII are underway in the USA and Europe.
Discussion
Since the description of the first patient with MPS VII by Sly et al in 19731 several authors have reported on the heterogeneous nature of the disease.4–9 ,39–44 Marked variation in phenotypical expression has been seen in the 90 patients with MPS VII reported in the literature and confirmed by the information obtained in this study. Including the patients from this study we have information on a total of 143 patients with MPS VII diagnosed in 30 countries. Overall, patients with mild or moderate manifestations have coarse facial features, corneal clouding, frequent upper respiratory infections but mild skeletal abnormalities. Patients with more severe phenotypes exhibit short stature and greater skeletal dysplasia, macrocephaly, recurrent ear infections, gingival hypertrophy, hepatosplenomegaly, hernias and cognitive impairment.4–7 ,9 ,39
The most severe form of MPS VII disease is characterised by the presence of NIHF. NIHF is a condition in which there is excessive fluid accumulation within fetal extravascular compartments and body cavities, that is not caused by red cell alloimmunisation.45 NIHF has been reported multiple times in fetuses with MPS VII showing marked ascites, oedema of the limbs, hepatosplenomegaly, delayed brain development, mild decrease of ventricular function, liver disease, severe lung hypoplasia, dilated heart and, at autopsy, presence of foamy macrophages in the brain.21 ,46–55 One of the suggested mechanisms to explain hydrops in some MPS VII cases is the obstructive effects of hepatic sinusoidal infiltration leading to generalised oedema.21 In our survey, 23 patients (41%) had a history of hydrops (neonatal NIHF or infantile). In addition, one patient with MPS VII was reported to have isolated neonatal ascites without generalised oedema.56 These results are similar to figures in the literature in which 35 out of 90 cases (38.8%) with MPS VII had a history of NIHF. Thus, it is important to include MPS VII in the differential diagnosis of NIHF.
Cardiovascular involvement has been reported in many patients with MPS VII. Findings included left ventricular hypertrophy, aortic insufficiency, mitral regurgitation, congestive cardiac failure, aortic stenosis, and thickened aortic and mitral valves.9 ,30 ,57–61 In this study over 50% of patients with MPS VII had cardiac valve disease. Indeed, the postmortem analysis of the first patient reported with MPS VII, who died suddenly at age 20 years, found extensive cardiovascular lesions including arterial stenosis and marked fibrous thickening of the atrioventricular and aortic valves.62 Similar cardiac findings at autopsy of another patient aged 28 years were recently reported.63
In our study we observed decreased pulmonary function (both restrictive and obstructive) in 71% of the patients. Thoracic deformities including pectus carinatum or excavatum contribute to restrictive airway disease leading to chronic hypoventilation with low forced vital capacity.61 ,64 There are reports of individual patients with severe pulmonary disease including bronchopulmonary dysplasia with fibrosis, recurrent pneumothoraces, interstitial lung disease and prolonged oxygen dependency.
In this study 90% of patients with MPS VII had skeletal dysplasia. Bone abnormalities in these patients and those previously reported included vertebral beaking, platyspondyly, hypoplastic odontoid process with atlantoaxial instability, kyphosis, scoliosis, pectus carinatum, bilateral dysplasia of femoral head and acetabulum, hip subluxation, widening of iliac wings, genu valgum, femoral capital epiphyses flattened and irregular, acetabular roofs irregular and fragmented, pseudoarthrosis, and interestingly, early bone maturation in utero.40 ,44 ,57 ,64–67
Skeletal changes often limit mobility. In this study, 57% of the patients (n=16) could walk without assistance. Twenty-one per cent of patients used a wheelchair, 11% used a walker or crutches and 11% could not mobilise at all. Though not frequently commented on in this study, odontoid hypoplasia has been reported in many patients.1 ,18 ,38 Its detection is important because atlantoaxial instability of the neck can pose a threat of cervical dislocation from trauma or intubation for anaesthesia. The patient shown in figure 1 died at age 12 years undergoing dental anaesthesia.
Central nervous system findings are prominent in patients with MPS VII. In our survey 28% of patients had cord-compression. Associated signs include hyper-reflexia and clonus. In addition, 94% had delay in their linguistic domain and 86% had mental retardation. Previous reports have also noted the presence of mental retardation and delay in cognitive, linguistic and social domains in patients with MPS VII.68 A postmortem analysis in a 20-year-old adult patient showed neuronal loss in substantia nigra and anterior columns of the spinal cord.62
Hearing impairment is reported to be common in patients with MPS VII.68 In this survey we found that 41% of patients with MPS VII had sensorineural hearing loss. Chronic otitis media and upper respiratory tract infections have been reported to be common.68 In this study 52% of the patients had ear infections and 69% had respiratory infections.
Other findings in occasional patients with MPS VII include kidney enlargement, renal insufficiency, hemiparesis, retinitis pigmentosa and some gastrointestinal (GI) disturbances including diarrhoea, dysphagia, reflux and colitis. Four patients had neonatal cholestasis (ref. 58, Bérubé et al; this study).
Phenotypical characteristics of patients with MPS VII resemble those of MPS I and MPS II (short stature, skeletal dysplasia, hepatosplenomegaly, hernias, cardiac involvement, decreased pulmonary function and cognitive impairment). In MPS VII, however, one unique and distinguishing clinical feature is the unexpectedly high proportion of patients (41%) that had a history of NIHF. Despite the prenatal onset of clinical disease in these patients, 13 out of 23 survived infancy with a mild to intermediate course into their late teens. Thus presence of NIHF does not, by itself, predict the eventual severity of the clinical course, if the patient survives infancy.
Causes of death in patients with MPS VII reported in the literature and in this study included cardiac arrhythmia, kidney failure, cardiac arrest, and in one case, pulmonary aspiration.26 ,44 ,62
For many years palliative therapies were the only resource for patients with MPS. These include surgeries, oxygen supplementation, antibiotics, non-steroidal anti-inflammatory drugs and physical therapy. Although specific treatments for MPS VII have lagged behind therapies for other LSDs because of its rarity and clinical variability, experimental approaches to treatment using animal models for this disorder have been very informative and have produced promising results.
The MPS VII mouse was shown to be greatly improved by ERT. The treatment effect was greatest if given early in life including prevention of some of learning, memory and hearing deficits.69 Vogler et al70 reported that high dose of enzyme (weekly injection of 4 mg/kg for 13 weeks) could cross the blood-brain barrier and improve glia and neocortical neurons. In addition, reports showed that chemically modified GUS enzyme which had longer circulating half-life improved clearance of GAG accumulation in several tissues including brain71 and skeletal tissue.72 BMT also altered the course of the disease and combination of ERT and BMT gave better results than either monotherapy.73 Gene therapy (GT) has been shown to be effective by several investigators. Davidson's group74 showed that GT directed to brain could reverse established cognitive impairment. Potentially promising results were also reported in an MPS VII dog using retroviral GT.75 These preclinical experiments in many laboratories provided optimism for the potential to develop ERT for patients with MPS VII. rhGUS is under clinical development as a potential ERT.
The first opportunity for clinical application was provided by a 12-year-old patient whose seriously deteriorating condition led to granting an emergency Investigational New Drug (IND) Application for ERT on a compassionate use basis. Results of this patient's response to therapy over the first 24 weeks were recently reported.38 Further studies documenting response over the 1st year were also recently presented.76 More recently a phase I/II clinical study was undertaken in UK involving 3 patients and a phase III study involving 12 patients in the USA is now underway.
Another approach to a non-palliative treatment is BMT. In BMT the enzyme that is missing is supplied endogenously through synthesis by the transplanted stem cells.77 Birkenmeier et al78 showed that BMT in adult GUS-deficient mice79 increased life span and produced clearance of GAGs in liver, spleen and partial correction in kidneys with minimal correction in the central nervous system and the skeleton.78 A Japanese patient underwent successful BMT at 12 years of age and after 2 years the levels of GUS were stable, with improvement of motor functions (walking, riding and taking bath alone), recurrent infections and snoring; however there was no reversal of the neurological damage.80 A Mexican female patient with MPS VII with genotype P408S/P415L underwent BMT at 11 years of age and was reported to be doing well 2 years after the transplantation.30 In the current study, five patients underwent BMT. Two of them died but the three survivors had some clinical benefit. The limited results suggest that BMT can slow or even prevent further neurological complications, but has little to no effect on the skeletal disease unless it is performed in neonates.81 ,82 The main limitations of BMT in MPS have been the difficulty in finding appropriate donors and the high morbidity and mortality after BMT. Availability of donors improved when using umbilical cord blood as a source of stem cell became accepted. Haematopoietic stem cells from umbilical cord human leukocyte antigen (HLA) system matched have proved to be safe and effective, are much more readily available than bone marrow, and require a less strict HLA match.77
Although MPS VII was the first MPS that had its enzyme characterised catalytically and the first to provide an ideal mouse model, it took over 40 years from the time of its discovery until the possibility of clinical trials could be realised. Along the way, studies of cells from the first identified patient had an important impact on the field. They led to the elucidation of the mannose-6 phosphate targeting system on which most of the ERTs are dependent. Studies of the clearance and uptake of the non-phosphorylated form of GUS led to elucidation of the mannose targeting system which formed the basis for ERT in Gaucher disease. Lessons from the mouse model of MPS VII have been similarly influential. The great leap from mouse to man is anxiously awaited.
Acknowledgments
The authors thank Anita Misra-Press for providing medical writing support.
References
Footnotes
Dr Tanaka died on 11 July 2015.
Contributors AMM designed the research study. AMM performed the interviews. NL-H, RDS, BHG, MS, RG, MP, AG-M, MÇ, DB, MSS, RW, RGi, AM, GP, AKK, FE, AT, LA, MB, REF, KB, JF, KKW, GAM, LC and WSS contributed clinical information. AMM and MH analysed the data. AMM and WSS wrote the manuscript with inputs from all the coauthors.
Funding This work was supported in part by Ultragenyx Pharmaceutical grant number 16232.
Competing interests AMM has received funds from Ultragenyx Pharmaceutical to conduct this survey. AG-M and RW are principal investigators in clinical trials in patients with MPS VII supported by Ultragenyx Pharmaceutical. KW has received funds from BioMarin Pharmaceuticals. AMM has received funds from Actelion Pharmaceuticals. KW had received speaker honoraria/travel from Genzyme Corporation. AMM and KW had received speaker honoraria/travel from BioMarin Pharmaceuticals.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data gathered resulted in this publication.