Article Text

Abstract
Background The aim of this work was to identify new genetic causes of Rett-like phenotypes using array comparative genomic hybridisation and a whole exome sequencing approach.
Methods and results We studied a cohort of 19 Portuguese patients (16 girls, 3 boys) with a clinical presentation significantly overlapping Rett syndrome (RTT). Genetic analysis included filtering of the single nucleotide variants and indels with preference for de novo, homozygous/compound heterozygous, or maternally inherited X linked variants. Examination by MRI and muscle biopsies was also performed. Pathogenic genomic imbalances were found in two patients (10.5%): an 18q21.2 deletion encompassing four exons of the TCF4 gene and a mosaic UPD of chromosome 3. Variants in genes previously implicated in neurodevelopmental disorders (NDD) were identified in six patients (32%): de novo variants in EEF1A2, STXBP1 and ZNF238 were found in three patients, maternally inherited X linked variants in SLC35A2, ZFX and SHROOM4 were detected in two male patients and one homozygous variant in EIF2B2 was detected in one patient. Variants were also detected in five novel NDD candidate genes (26%): we identified de novo variants in the RHOBTB2, SMARCA1 and GABBR2 genes; a homozygous variant in EIF4G1; compound heterozygous variant in HTT.
Conclusions Network analysis reveals that these genes interact by means of protein interactions with each other and with the known RTT genes. These findings expand the phenotypical spectrum of previously known NDD genes to encompass RTT-like clinical presentations and identify new candidate genes for RTT-like phenotypes.
- Rett syndrome
- Epilepsy
- Whole exome sequencing
- Intellectual disability
Statistics from Altmetric.com
Introduction
Rett syndrome (RTT) is a severe neurodevelopmental disorder (NDD) affecting mostly girls, characterised by an apparently normal prenatal and perinatal period followed by a stagnation in development and a severe regression in language and motor skills.1 RTT is clinically divided into classical and atypical forms of the disease.2 The clinical diagnostic criteria for RTT can be revisited in table 1. Patients with RTT or RTT-like clinical presentation often present with severe intellectual disability (ID), autistic features and epilepsy, and their differential diagnosis includes Angelman syndrome, Pitt-Hopkins syndrome (PTHS) and some epileptic encephalopathies.3–5
Clinical diagnostic criteria for Rett syndrome (adapted from Neul and colleagues1)
Whole exome sequencing (WES), has had a major impact in medical practice, leading to the identification of several new genes involved in ID.6–8 We used a genomic approach combining array comparative genomic hybridisation (aCGH) and WES to find genetic causes of disease in a group of RTT-like patients who tested negative for MECP2 mutations and—whenever clinically appropriate—CDKL5 mutations. We were able to detect pathogenic variants and very likely pathogenic variants that we believe can account for the RTT-like phenotype in 13 (68%) of these patients.
Methods
Patients
We enrolled 19 patients (16 girls and 3 boys) with idiopathic neurodevelopmental phenotypes that clinically overlap with RTT and their unaffected parents (trios). The patients were selected from a previously established database of patients with idiopathic ID and confirmed as eligible by consulting with medical geneticists, paediatric neurologists and neurodevelopmental paediatricians, using the revised clinical criteria for RTT diagnosis.1 We included patients meeting sufficient criteria for the diagnosis of Rett (classical or atypical)—except for documented regression, which was not considered mandatory. Exclusion criteria were also taken into account (tables 1 and 2; online supplementary data1).
Clinical characterisation of the cohort relative to RTT clinical diagnosis criteria
Before enrolment all patients had undergone routine diagnostic workup, including brain MRI and metabolic screen. MECP2 analysis was performed by Sanger sequencing and qPCR for all patients and CDKL5 sequencing was undertaken for patients presenting early onset seizure variant. No patient presented with clearly congenital forms, hence FOXG1 was not tested. Patients would only be enrolled in the study if their complementary exams had been normal or with abnormalities that could not clearly explain the phenotype.
Molecular analysis
For all patients included in this work an aCGH analysis was performed first, followed by WES (provided aCGH profile had been normal or inconclusive).
Array comparative genomic hybridisation
aCGH analysis was performed using two different platforms: human genome CGH Agilent 180K custom array and Illumina HumanOmniExpress beadchip array (see online supplementary part 2; figure S2.1). All genomic coordinates are in build GRCh37/hg19.
Exome sequencing and variant detection
Exomes were enriched with Agilent's SureSelect All Human Exome V.4 Kit (51 Mb encompassing the exons of 20 965 genes), followed by AB SOLiD5500xl System sequencing (Life Technologies). Filtering of single nucleotide variants and indels is described in online supplementary data2. Preference was given to (1) de novo variants, (2) homozygous or compound heterozygous variants compatible with an autosomal recessive mode of transmission and (3) X linked variants. The impact of variants was predicted using in silico tools, namely SIFT,9 PolyPhen2,10 Mutation Assessor,11 Mutation Taster,12 PMut13 and Condel.14 Alignment for amino acid conservation among species was performed using the ClustalW2 webtool (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (see online supplementary data2; figure S2.2).
Selection and interpretation of the variants
Candidate variants were validated by Sanger sequencing in the trios. Variants were selected for Sanger confirmation as described in online supplementary data2 and in figure S2.3. The variants selected for Sanger sequencing confirmation are described in online supplementary data1, table S1.19. The primers designed for this purpose are listed in online supplementary data2, table S2.2. The variants were classified according to the flow chart depicted in online supplementary figure S2.4, adapted from de Ligt and colleagues.6
Network analysis
We performed gene network analysis to: (1) verify if our candidate genes interacted among themselves and with the known RTT genes (MECP2, CDKL5, FOXG1), (2) study the topology of these interactions, (3) predict additional genes that may be involved in RTT if they are shown to interact with a large number of genes in the query set, (4) identify common biological themes by exploring functional enrichment analysis of Gene Ontology (GO) terms.
Network analysis was performed with GeneMANIA (V.3.1.2.7, http://www.genemania.org/).15 ,16 Given a set of input genes, GeneMANIA finds related genes using a very large set of functional association data, including protein interactions, genetic interactions, pathway, coexpression, colocalisation, shared protein domain and predicted functional relationship. GeneMANIA also allows for functional enrichment analysis. For our analysis, the genes used as input were the already known RTT genes (MECP, CDKL5, FOXG1) as well as the genes selected as likely causing RTT-like phenotype in our cohort.
For additional details on the methodology of the gene network analysis performed using GeneMANIA (V.3.1.2.7) see online supplementary data2.15 ,16
Results
Patients' clinical profile
We enrolled 19 patients (16 girls and 3 boys) with ages between 6 and 31 years (mean age 15.8±6.3 years), with idiopathic neurodevelopmental phenotypes that clinically overlapped with RTT, as well as their unaffected parents (trios). The patients were selected from a previously established database of patients with ID and confirmed as eligible using the revised clinical criteria for RTT diagnosis.1 All patients had normal routine diagnostic workup, including brain MRI, which for RTT diagnosis purpose was classified as ‘normal’ if the alterations present were not a consequence of a perinatal or postnatal insult, neurometabolic disease or severe infection; this was the case for patients 1, 2, 6, 14, 15 and 17. Detailed description of the MRI findings for these patients can be found in the online supplementary data. Metabolic screen, MECP2 analysis by Sanger sequencing and qPCR, as well as CDKL5 sequencing (if early onset seizure variant) were performed for all patients.
The main clinical findings of the patients are summarised in table 2 (more detailed in online supplementary data1). We have classified the patients according to Neul and colleagues, as: (1) classical RTT, if they had had regression and presented all 4 main criteria; (2) atypical RTT, if they had had regression and met at least 2 of the 4 main criteria and 5 of the 11 minor criteria, and (3) RTT-like, if they met criteria for RTT (classical or atypical) except for documented regression. The most common comorbidities in this cohort were epilepsy, affecting 74% (14 of 19), and autism spectrum disorder (ASD) in 74% (14 of 19) of the patients. All patients were sporadic cases and no other relevant findings were reported in their family history, except where specified below. The clinical features of the cohort are summarised in table 2.
Global yield of genomic analysis
Using the combined aCGH and WES analysis we were able to detect genomic imbalances in 10.5% (2 out of 19) of the patients and single nucleotide variants in 58% (11 out of 19) patients (summary of the results in online supplementary table S1.1 and interpretation workflow in online supplementary figure S2.1). We detected a de novo 18q21.1 microdeletion encompassing the TCF4 gene in patient 7, which was confirmed by qPCR, and a mosaic UPD of chromosome 3 in patient 16 (see online supplementary figures S1.1 and S1.2, respectively). For patients without diagnostic findings in the aCGH analysis, we performed WES analysis (see online supplementary figure S2.2–S2.4). A summary of the WES results is provided in tables 3 and 4: six variants in six genes previously described as associated with NDDs (table 3); likely pathogenic variants in five genes not described as associated with a RTT-like or ID related phenotype but which, due to their functions, may account for the disease in the patients (table 4).
List of patients with variants found in genes previously associated with neurodevelopmental phenotype
List of patients with variants in candidate disease genes
Patients with CNVs causing RTT-like phenotypes
Patient 7 is a girl who had an apparently normal development up until the age of 4 months, when regression was noticed. Hand stereotypies were documented around 30 months; still, acquisition of some hand skills occurred around 6 years of age, but these were lost 3 years later. Currently the patient is an adult with moderate ID, epilepsy and autistic behaviour. Additionally, she presents with eight minor criteria for RTT diagnosis, including respiratory disturbances—more precisely hyperventilation. Overall, the patient was classified as atypical RTT. Microarrays revealed a de novo 0, 25 Mb microdeletion at chromosome 18q21.1 encompassing four exons of TCF4 and the MIR4529 gene (see online supplementary figure S1.1). Loss of function mutations and microdeletions affecting TCF4 have been described in patients with PTHS.19 ,20 ,34 The natural history of RTT and PTHS overlaps significantly, the latter being usually considered in the differential diagnosis of RTT. The fact that the patient is a girl, lacks dysmorphisms, started to show stereotypies around 2.5 years old and hyperpnoea at 7 years lead to the consideration of RTT as a first possibility.
Patient 16 is a 9 year-old girl who showed developmental stagnation at around 6 months, which coincided with appearance of West syndrome and deceleration of head growth. She has slowly acquired motor skills, with some purposeful grasp and ataxic gait. Severe ID and hand wringing raised the diagnostic hypothesis of RTT. aCGH analysis revealed an entire chromosome 3 with log R ratio (LRR)=0 and B Allele Frequency (BAF) split (0.3 and 0.6), compatible with mosaic UPD of chromosome 3 (see online supplementary figure S1.2). This abnormality occurred de novo and is predicted to be present in about 75% of the cells of the patient.35 Only three of all the genes in chromosome 3 are predicted to be differently expressed according to parental lineage: ALDH1L1 and ZIC1 are paternally imprinted, and HES1 is maternally imprinted.36 When considering the possibility of a variant in heterozygosity in the mother/father being present in homozygosity in the child's cells with UPD, only a maternal missense variant in SRGAP3 found in the WES analysis fits this hypothesis. Interestingly, SRGAP3 encodes a Slit-Robo Rho GTPase activating protein that has been implicated in the pathogenesis of ID.37 ,38
Patients with variants in genes previously associated with similar phenotype
Patient 5 is a 5 year-old girl who regressed at around 8 months of life. Though the child presents with severe ID and no language, some psychomotor developmental milestones were attained, with tiptoe walking around 2 years of age. Hand stereotypies, intense eye communication, breathing disturbances (apnoea followed by hyperpnoea) and screaming spells prompted the clinical diagnosis of atypical RTT. Though severely microcephalic, the patient's brain MRI did not reveal any relevant morphological changes. Also, the patient does not have short stature. In this patient, a de novo c.C556T, p.(R186X) variant was detected in the ZNF238 gene (see online supplementary figure S1.5).
Patient 17 is a 6 year-old girl who presented with seizures at 1 month of life and whose development was significantly delayed, with first words around 3 years and walking at 4 years of age. The patient also has hand stereotypies, bruxism and crying spells when awake, sleep problems, hyperpnoeas and apnoeas, and poor eye contact. In this patient a de novo c.G274A, p.(A92T) substitution in the EEF1A2 gene was found (see online supplementary figure S1.15).
Patient 8 is a 13 year-old girl and the first child of a healthy and non-consanguineous couple. She seemed to have a normal development up until 24 months when regression was noticed in language and hand manipulation, followed by the first signs of autistic behaviour-like impairment in social interactions around 30 months. Concomitant hand stereotypies, ataxia, rigidity and screaming spells led to the classification of the patient as RTT-like. The patient also presented continuous tremor but no spasticity or optic atrophy. Brain MRI performed when the patient was 3 years old did not reveal significant alterations. In this patient a homozygous recessive variant c.C380T, p.(A127V) in the EIF2B2 gene was found (see online supplementary figure S1.7).
Patient 14 is a 12 year-old boy who presented with West syndrome at 1 month of age and severe developmental delay, in addition to poor eye contact, hyperventilation episodes, bruxism when awake, decreased response to pain and midline hand stereotypies. This RTT-like patient has a de novo c.T538C, p.(C180M) variant in the STXBP1 gene (see online supplementary figure S1.13). A missense mutation in amino acid 180 was described in 2008.39 Although variable,40 the core phenotype of STXBP1 mutations seems to include epilepsy within the 1st months of life.41 Mutations in STXBP1 can also cause ID without epilepsy22 and it may actually be a relatively common cause of severe ID,42 which highlights the role of STXBP1 in cognitive function alone. Recently, patients with STXBP1 mutations were noted to have head and upper limb stereotypies (eg, hand flapping)43 but midline hand stereotypies are reported for the first time in our study. While our manuscript was in preparation a de novo missense variant in STXBP1 was reported in a girl with classical Rett diagnosis.44
Patient 15 is an 8 year-old boy whose development stagnated around 6 months; his psychomotor development was significantly delayed, language and gait never being attained. The patient also had an abnormal sleep pattern, inconsolable crying spells and hand and head stereotypies, as well as autism features; he was classified as RTT-like. WES revealed a c.G772A, p.(V258M) variant in the SLC35A2 gene (see online supplementary figure S1.14). The patient’s mother also carried the variant and presented with a random X inactivation. The variant was also present in the patient’s skin biopsy. Transferrin isoelectric focusing analysis in the patient was normal. Interestingly, upon reanalysis of the clinical data, we found that the patient also had facial dysmorphisms, gastro-oesophageal reflux, epileptic encephalopathy (but not West syndrome), microcephaly and brain malformations (namely brain atrophy, thin corpus callosum and frontoparietal periventricular heterotopies), resembling other patients with SLC35A2 variants.23 ,24
Patient 19 is 14 year-old boy with dyspraxic gait and no language, who acquired purposeful grasp only around 2 years of age. Eye pointing, kyphosis, peripheral vasomotor disturbances and small cold hand and feet lead to the classification as RTT-like. This patient carries two maternally inherited X linked variants: a c.C436T, p.(R146W) variant in SHROOM4 and a c.G409A, p.(D137N) variant in ZFX (see online supplementary figures S1.17 and S1.18). Extended pedigree analysis reveals that patient 19 is an isolated case and both variants are inherited from the healthy mother, who has a random X inactivation pattern. SHROOM4 encodes a regulator of cytoskeletal architecture and has been associated with X linked ID.26
Other variants in genes previously associated with NDDs but which weren’t in accordance with the inheritance patterns described in the literature were also found in some cases (see online supplementary data1, tables S1.2–S1.18).
Patients with variants in genes possibly relevant for ID pathogenesis
For five of the patients enrolled in the study, likely pathogenic variants were found in functionally relevant and/or candidate ID genes.
Patient 2 is a 18 year-old girl who showed developmental regression around 6 months of life and 2 months later started to have partial complex seizures as well as lack of interest in interacting with the environment. This classical RTT patient meets all four main criteria in addition to eight supportive criteria. On neurological exam it was also observed that the patient had swallowing problems, dystonia and bradykinesia (but not rigidity) in addition to continuous manual stereotypies (but not chorea). Interestingly, brain MRI performed when the patient was approximately 5 years old showed significant striatum atrophy (especially in the caudate nuclei) as well as mild atrophy of the cortex and cerebellar vermis. WES revealed two compound heterozygous variants in the HTT gene (see online supplementary figure S1.3): a maternal c.C2108T, p.(P703L) and a paternal c.C3779T, p.(T1260M). The latter variant is described in single nucleotide polymorphism database (dbSNP) as a polymorphism (rs34315806) with a minor allele frequency of T=0.0276/138.
Patient 4 is a girl who has been classified as classical RTT. She had moderate developmental delay, superimposed by regression at around 5 years of age. Currently, at 25 years of age, the patient is severely autistic, non-verbal, can't walk, has lost purposeful hand use and has hand sterotypies, in addition to seven minor criteria (see details in table 2). She carries a de novo variant c.G2897T, p.(G966V) in SMARCA1 (see online supplementary figure S1.4).
Patient 9 had developmental stagnation at 7 months followed by regression, currently (at age 19 years) presenting with severe ID. She was classified as an RTT variant in light of her absence of language, hand stereotypies and lack of hand use, breathing disturbances (hyperventilation), bruxism, abnormal sleep cycle, crying spells, autistic features, eye pointing and small feet. She has never had seizures. A de novo variant c.G1699A, p.(A567T) in the GABBR2 gene was found (see online supplementary figure S1.8).
Patient 11 is a 6 year-old girl whose development stagnated at around 6–9 months, coinciding with the beginning of generalised epilepsy. Additional findings that lead to the classification of the patient as RTT-like include: hand stereotypies, intense eye communication, sleep problems, peripheral vasomotor disturbances, bruxism when awake, growth retardation, diminished response to pain and resting tremor. Her mother has resting tremor and is suspected of having psychiatric disease, possibly early onset dementia. The maternal grandmother is bedridden and demented. The father is also suspected of having psychiatric disease. The patient carries a de novo variant c.A1528G, p.(N510D) in RHOBTB2 (see online supplementary figure S1.10). She also has a homozygous c.G602A, p.(R201H) variant in EIF4G1 (see online supplementary figure S1.11).
Bioinformatic analysis of the interactions between the novel candidate genes and known RTT-like NDD associated genes
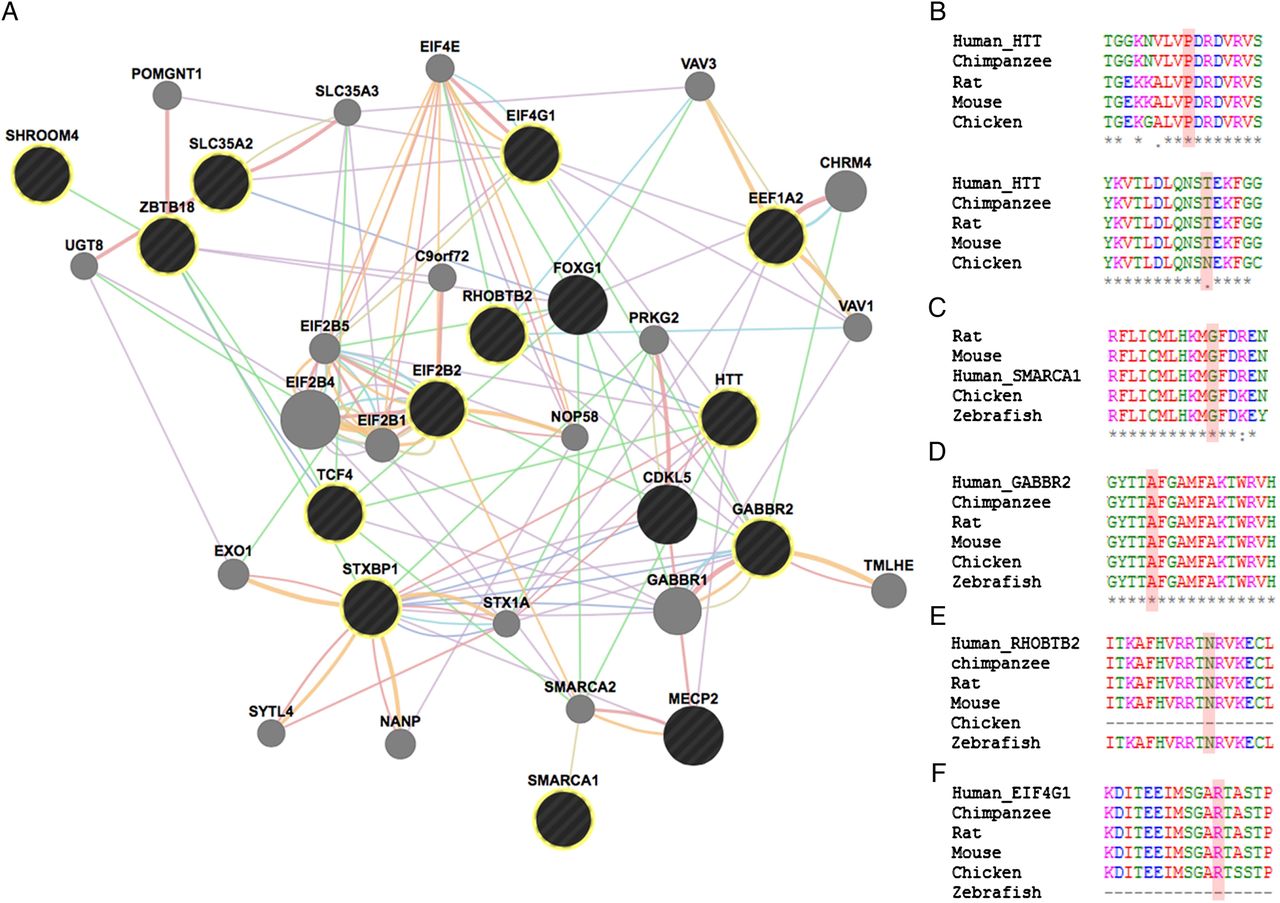
Phenotypical overlap between RTT and the patients in our cohort with variants in genes previously implicated in NDDs was observed (table 2 and figure 1). Network analysis using GeneMania revealed that our candidate genes interact with each other and with the already known RTT genes by means of protein (71%), predicted (15%) and genetic (3%) interactions, as well as coexpression (7%) and participating in a common pathway (2%) (figure 2).
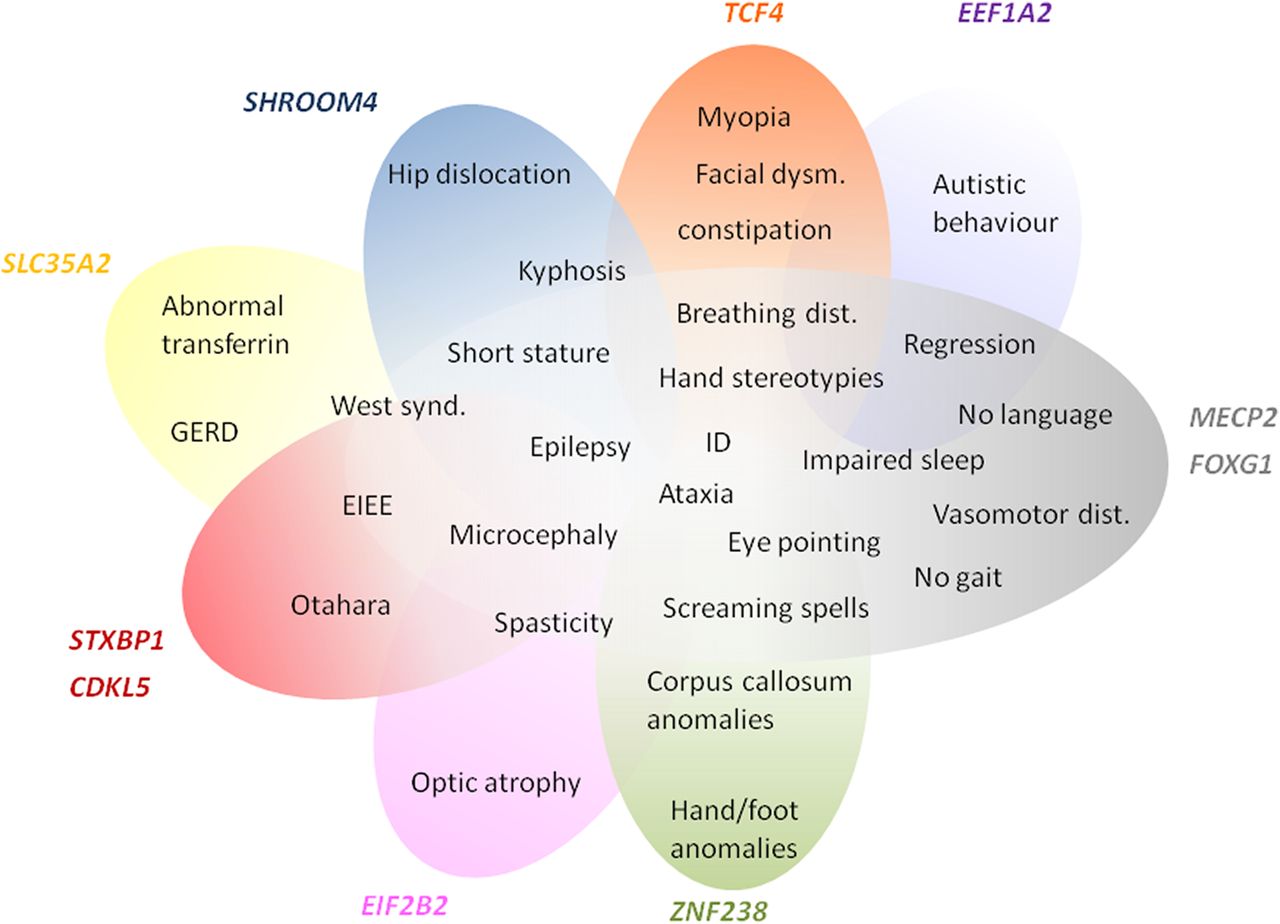
Scheme representing phenotype overlap of the genes already identified as causing neurodevelopmental disorders and the RTT phenotype. The phenotypes clearly blend, suggesting that the RTT spectrum may still be expanding.

{kind=link}
{kind=link}
(A) Functional network showing the interaction among genes with variants in our cohort and genes previously associated with RTT. GeneMANIA retrieved interactions between the query genes (black circles—highlighted with yellow circumference if identified in our cohort) and added extra genes (grey circles) that are strongly connected to the query genes. Analysis was based on physical interactions (red edges), predicted interactions (orange edges), coexpression (purple edges), genetic interactions (green edges), pathway (light blue edges), colocalisation (dark blue edges) and shared protein domains (brown edges). (B) Species alignment for the changed animo acid for Htt p.(P703L) (above) and Htt p.(T1260M) (bellow), (C) Smarca1 p.(G966V), (D) Gabbr2 p.(A567T), (E) Rhobtb2 p.(N510D) and (F) Eif4g1 p.(R201H).
Discussion
In this study we identified a possible genetic cause of disease in eight RTT-like patients. Recently, two publications described the application of WES to smaller series of patients with features of RTT, where, if we exclude the variants found in MECP2 and FOXG1 genes, an yield of 27% was found.45 ,46 However, important questions such as (1) what other genes may lead to an RTT-like similar phenotype and (2) which pathways and genetic mechanisms can lead to such a specific phenotype still remain unanswered. To try to clarify these questions we undertook genomic analysis by aCGH and WES in a group of 19 trios whose index presented NDD with RTT-like features achieving an yield of 37% (excluding a case of uniparental dissomy and variants found in candidate genes).
Regarding the WES results, in 6 of the 19 patients we detected variants in genes previously associated with overlapping neurodevelopmental phenotypes, namely: SLC35A2, STXBP1, ZNF238, EEF1A2, EIF2B2 and SHROOM4 (table 3). On the other hand, five patients present novel variants in genes with known function in the central nervous system and which, to the best of our knowledge, have not been clearly associated with ID in humans (HTT, SMARCA1, GABBR2, RHOBTB2 and EIF4G1) (table 4). All these genes, taking into account their function, are good candidates to be disease-causing. HTT encodes a protein that directly interacts with MeCP227 and Mecp2-deficient mice have reduced expression of Htt in the entire brain, leading to a defect in the axonal transport of Bdnf.28 RTT and Huntington disease seem to share features at a molecular level (nuclear interaction for transcriptional regulation and axonal trafficking through BDNF)27 and neuropathological findings (striatum atrophy)47 and clinical presentation (of compulsive movement disorder plus cognitive dysfunction).
Another of our candidates, SMARCA1 (alias SNF2L), encodes a protein which was described to function antagonistically with Foxg1 in the regulation of brain size in mice.29 ,48
GABBR2 encodes a γ-aminobutyric acid type B receptor that is involved in neuronal activity inhibition through the regulation of neurotransmitter release.31 Recently, de novo missense variants in GABRR2 were identified in two different patients with infantile spasms.49
RHOBTB2 belongs to the Rho GTPases family and was found to bind to CUL3.32 De novo nonsense variants in CUL3 were identified in two separate next-generation sequencing studies using ASD (autism spectrum disorder) probands,50 ,51 EIF4G1 encodes a translation initiation factor involved in the recruitment of mRNA to the 40S ribosomal subunit.33 Variants in EIF4G1 have been associated with autosomal dominant forms of Lewy body dementia52 and Parkinson's disease (with and without dementia) however the real impact of some of these variants is still unclear.53–56 Interestingly patient 11 (homozygous) and her mother (heterozygous) present resting tremor, a typical sign of Parkinson's disease. Considering the biological processes in which RHOBTB2 and EIF4G1 are involved, it is likely that either one of the variants or the combination of both in the patient might have contributed towards the development of the disease.
When analysing the phenotypes associated with genes previously implicated in NDDs, they overlap with those observed in our sample of RTT-like patients. In fact, there seems to be a spectrum of clinical presentation that allows for the delineation of a core phenotype as well as distinctive clinical features, that could help guide/interpret genetic testing in future patients, as summarised in figure 1. Furthermore, the gene-gene interaction analysis revealed that our candidate genes interact with each other and with the already known RTT genes mainly by means of protein interactions (71%), predicted functional relationships (15%) and coexpression (7%) (figure 2).16 Functional enrichment analysis revealed that the top GO biological process terms were under the parent terms Translation (GO:0006412) and Glial cell differentiation (GO:0010001). Careful analysis of the network also allows for identification of possible additional candidates such as GABBR1, which has genetic interactions with FOXG1, physical interactions with GABBR2 and coexpression with STXBP1 and TCF4.
In this work, the identification of variants in genes that had already been associated with overlapping but still distinctive NDDs brings new insight into the differential diagnosis of RTT and might allow for the aetiological diagnosis of RTT-like patients. We point out seven novel candidate genes, which may be implicated in RTT-like clinical presentations. It is important to highlight that replication of these results in more patients is required for a proper genotype-phenotype correlation and the establishment of differences and similarities with RTT. Functional studies would also be of great value. In conclusion, we expanded the phenotypical spectrum of previously known NDD genes to encompass RTT-like clinical features, and suggest novel genes that might be associated with those. Although this group of disorders is genetically heterogeneous, the novel and previously identified genes converge in common pathways and only a better understanding of the pathophysiology of NDDs will allow for development of efficient targeted therapies in the future.
Acknowledgments
The authors thank the patients and families for participation in the genetic studies and for allowing this publication. SOLiD sequencing was performed at the Uppsala node of the National Genomics Infrastructure (NGI) Sweden and computational resources from Uppnex were used for the data analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
FL and MB contributed equally.
Contributors FL and MB: laboratory work, acquisition of data, analysis, interpretation of results and manuscript preparation; AA: bioinformatics and statistical analysis; GS: recruitment of patients, acquisition and analysis of clinical data and manuscript preparation; JdS, AID, GO, PC, TT, EC, IFC, JPV, RO: recruitment of patients and acquisition of clinical data; SE: in silico tools analysis; IJ, A-CS, UG: study supervision; DP: aCGH analysis; PM: study concept and design, data interpretation, critical revision of the manuscript for important intellectual content, study supervision.
Funding This work was supported by the Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 262055. This work was also supported by the FEDER through the Programa Operacional Factores de Competitividade—COMPETE and by Portuguese national funds through the FCT—Fundação para a Ciência e Tecnologia, grants number PIC/IC/83026/2007 and PIC/IC/83013/2007, PhD scholarship grant to MB number SFRH/BDINT/ 51549/2011 and PhD scholarship grant to FL number SFRH/BD/84650/2010.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Hospital Geral de St. António, Portugal.
Provenance and peer review Not commissioned; externally peer reviewed.