Article Text

Abstract
Background SOX11 is a transcription factor proposed to play a role in brain development. The relevance of SOX11 to human developmental disorders was suggested by a recent report of SOX11 mutations in two patients with Coffin–Siris syndrome. Here we further investigate the role of SOX11 variants in neurodevelopmental disorders.
Methods We used array based comparative genomic hybridisation and trio exome sequencing to identify children with intellectual disability who have deletions or de novo point mutations disrupting SOX11. The pathogenicity of the SOX11 mutations was assessed using an in vitro gene expression reporter system. Loss-of-function experiments were performed in xenopus by knockdown of Sox11 expression.
Results We identified seven individuals with chromosome 2p25 deletions involving SOX11. Trio exome sequencing identified three de novo SOX11 variants, two missense (p.K50N; p.P120H) and one nonsense (p.C29*). The biological consequences of the missense mutations were assessed using an in vitro gene expression system. These individuals had microcephaly, developmental delay and shared dysmorphic features compatible with mild Coffin–Siris syndrome. To further investigate the function of SOX11, we knocked down the orthologous gene in xenopus. Morphants had significant reduction in head size compared with controls. This suggests that SOX11 loss of function can be associated with microcephaly.
Conclusions We thus propose that SOX11 deletion or mutation can present with a Coffin–Siris phenotype.
- Developmental
- Clinical genetics
- Copy-number
- Diagnostics tests
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
The SOX proteins are transcription factors with a shared motif called the SRY box, a high mobility group (HMG) DNA binding domain. The SOX proteins regulate gene expression, acting as either transcriptional activators or repressors, in multiple tissues, and so play crucial roles in multiple developmental processes.1 SOX11 is thought to play a crucial role in brain development. In humans, neuron production begins on embryonic day 42.2 In the fetus, the neuronal progenitors are located in the subventricular zone. After production in the subventricular zone, neurons migrate outwards into the cortical layers and undergo differentiation into mature neurons. The linked processes of neuronal production from progenitor cells and differentiation into functioning neurons must be tightly regulated to ensure proper brain development.2 SOX11 null mice have reduced cortical neurogenesis secondary to reduced proliferation and abnormal differentiation of neuronal progenitor cells.3 This results in SOX11 null mice having reduced brain weights and thin cerebral cortices.3 There is also evidence that SOX11 plays a role in ocular development. Sox11 knockdown in zebrafish induces microphthalmia with or without iris coloboma.4 SOX11 represents a strong candidate gene for human neurodevelopmental disease.
Haploinsufficiency of other SOX genes is associated with human disease. Mutations in SOX10 are associated with Waardenburg–Hirschprung disease,5 SOX9 mutations with campomelic dysplasia6 and haploinsufficiency of SOX5 is reported to cause intellectual disability.7 Tsurusaki et al8 reported two children with Coffin–Siris syndrome (CSS, OMIM#135900) and de novo mutations in SOX11. CSS is characterised by developmental delay/intellectual disability, feeding difficulties, facial dysmorphology, microcephaly and hypoplastic nails of the fifth digits.9 Both of the mutations reported by Tsurusaki et al8 were within the HMG domain and interfered with the ability of SOX11 to induce gene transcription in vitro. This implicates regulation of gene expression as a mechanism by which SOX11 contributes to human brain development. Multiple genes regulated at a transcriptional level by SOX11 have been identified.10 SOX11 can also repress transcription of genes important for neurodevelopment. In SOX11 null mice, LIS1 was upregulated significantly.3 Altered levels of SOX11 thus have the potential to cause dysregulation of multiple genetic pathways, with clear potential to disrupt developmental processes.
Here we report seven individuals with chromosome 2p25 deletions including SOX11 and three with de novo SOX11 mutations. These individuals presented with a phenotype that had some clinical features of CSS, but not a classical phenotype that would readily permit clinical diagnosis of CSS. An in silico analysis demonstrated that expression of SOX11 is highest in the brain during early fetal life, suggesting a role for SOX11 in human neurodevelopment. Knockdown of Sox11 in xenopus laevis was associated with microcephaly in the morphants.
Materials and methods
Ascertainment of SOX11 deletion (2p25.2 deletions) and mutation cases
Individuals with deletion of chromosome 2p25.2, which included the SOX11 gene, were identified through the DECIPHER collaboration. Deletions were confirmed by FISH. None of the deletions identified were present in the recently published CNV map of the human genome, which integrates CNV data from healthy individuals from multiple data sets such as the database of genomic variants.11 Two individuals with SOX11 mutations were identified in the deciphering developmental disorders (DDD) study (data freeze of 1133 children). DDD methodology has been described.12 A third individual with a SOX11 mutation was identified by exome sequencing via the Genetics of Structural Brain Abnormalities and Learning Disabilities Study (Wales Research Ethics Committee 12/WA/0001).13 Mutations were confirmed by Sanger sequencing.
In silico assessment of pathogenicity of novel SOX11 mutations
The predicted effect of the SOX11 missense variants was examined using SIFT, PolyPhen and the ‘Have Your Protein Explained’ tool (http://www.cmbi.ru.nl/hope/home). Evolutionary conservation of mutated amino acids was assessed by aligning orthologues in Ensembl (http://www.ensembl.org/index.html). The presence of SOX11 variants in normal control populations was queried using the ExAC browser (http://exac.broadinstitute.org/gene/ENSG00000176887).
Cell transfection and luciferase reporter assays
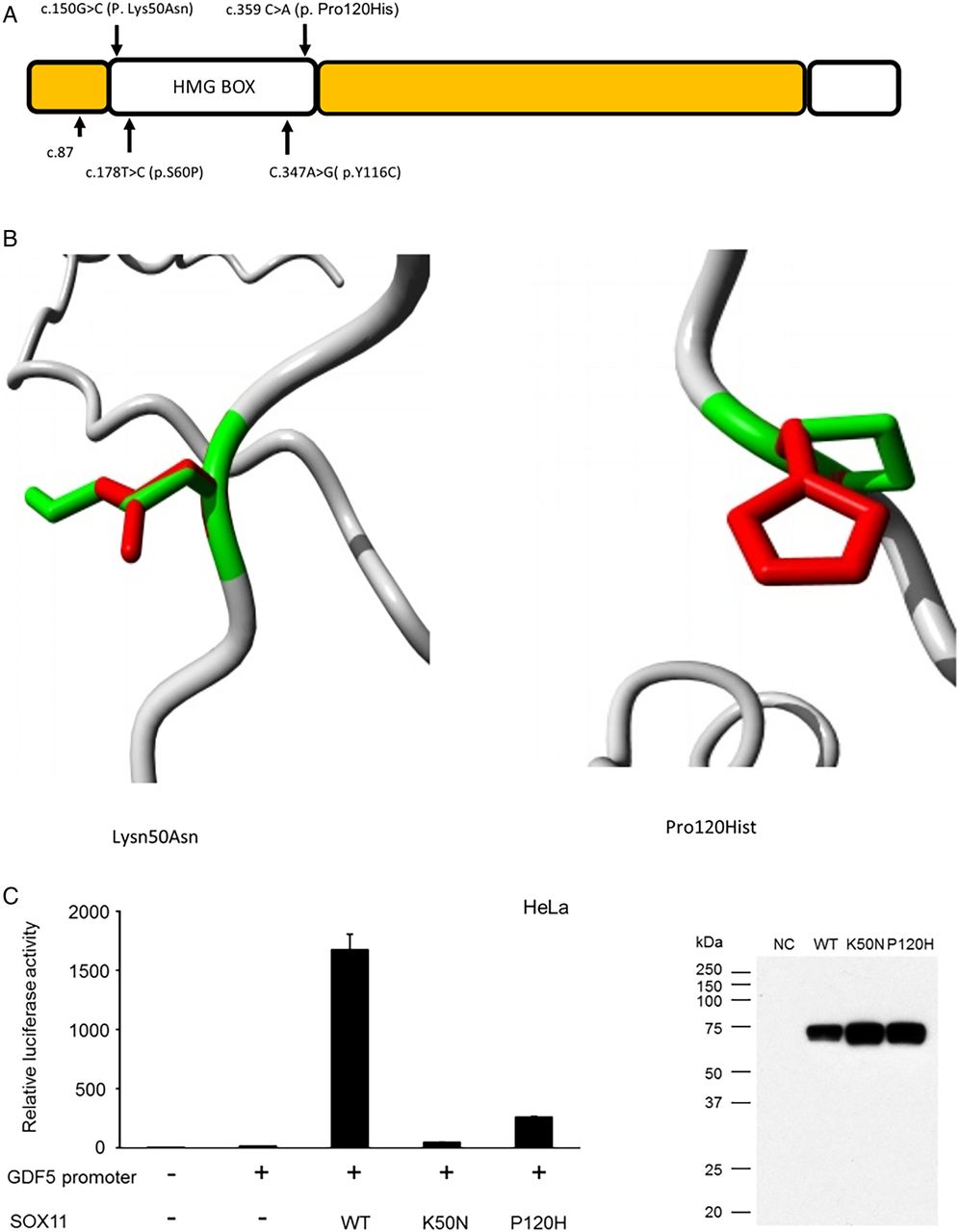
The SOX11 open-reading frame clone was purchased from Promega (Tokyo, Japan) and SOX11 mutants (c. 150G>C; p. Lys50Asn and c.359C>A; p. Pro120His) generated by site-directed mutagenesis with the KOD-Plus-Mutagenesis Kit (Toyobo, Osaka, Japan). Wildtype (WT) and mutant SOX11 cDNAs were PCR amplified and cloned into the p3xFLAG-CMV-14 mammalian expression vector (Sigma, St Louis, Missouri, USA). The GDF5 promoter 5′-flanking sequence (−448/+319) was PCR amplified and cloned into the pGL3-basic vector (Promega). All constructs were verified by Sanger sequencing. Human SOX11 cDNA can be obtained from GenBank/EMBL/DDBJ nucleotide core database under the accession code AB028641.1. Transfection and luciferase reporter assays were performed as previously described.8
In silico assessment of SOX11 expression in developing brain
Variation of SOX11 expression levels in the human brain over different developmental stages was investigated using RNA-sequencing data from the Brainspan atlas of the developing human brain (http://www.brainspan.org/). Methods are described in the online supplementary methods.
Sox11 knockdown in xenopus laevis embryos
Xenopus laevis embryos were obtained and cultured according to standard protocols and staged as described previously.14–16 All morpholino oligonucleotides (MOs) were obtained by GeneTools, LLC, OR, USA, and resuspended in Diethylpyrocarbonate (DEPC)-treated water. For loss-of-function experiments, Sox11 MO (30 ng per blastomere) was injected.14 For control experiments, the standard control MO suggested by GeneTools was used. MOs were injected bilaterally into both dorso-animal blastomeres of Xenopus embryos at eight-cell stage to target anterior neural tissue. As a lineage tracer, 0.5 ng gfp RNA was co-injected in all experiments to ensure proper injections. For cephalic evaluations, Xenopus embryos at stage 45 were fixed with formaldehyde and imaged using a Zeiss Axiophot microscope. Head area and interpupillary distance in knockdown and control morphants was compared using the Mann–Whitney U test (GraphPad prism).
Results
Clinical case reports
Individuals with SOX11 deletions
Clinical details, deletion mapping and photographs are given in table 1 and figures 1 and 2. Case 1 is a 12-year-old girl, the first child of healthy non-consanguineous parents (previously described at 7 years old17). She was born at 41 weeks of gestation by caesarean section due to fetal bradycardia and oligohydramnios. Birth weight was 3685 g (50th centile), length 50 cm (50th centile) and birth head circumference 35 cm (50th centile). At the age of 12 years, her head circumference was 50.4 cm (<3rd centile), height 152 cm (25–50th) and weight 44 kg (75th centile). She walked without support at 18 months and has poor fine motor skills. She started speaking single words at the age of 3 years and 6 months and has had no further language development. She manifests repetitive and stereotyped movements with hyperactivity and autism. She had a non-verbal IQ of 54. She was generally well apart from gastro-oesophageal reflux. On examination she has mild facial asymmetry with right microphthalmia and a wide mouth with thick lips and large, simplified ears. Bilateral fifth finger clinodactyly, cutaneous syndactyly of toes 2–3, scoliosis and inverted nipples were also noted. CGH was reported as arr 2p25(4291420–6905655)(hg 19)x1 with deletion of the SOX11 gene only. The deletion was not present in either parent.
Summary of demographic, genetic and clinical characteristics
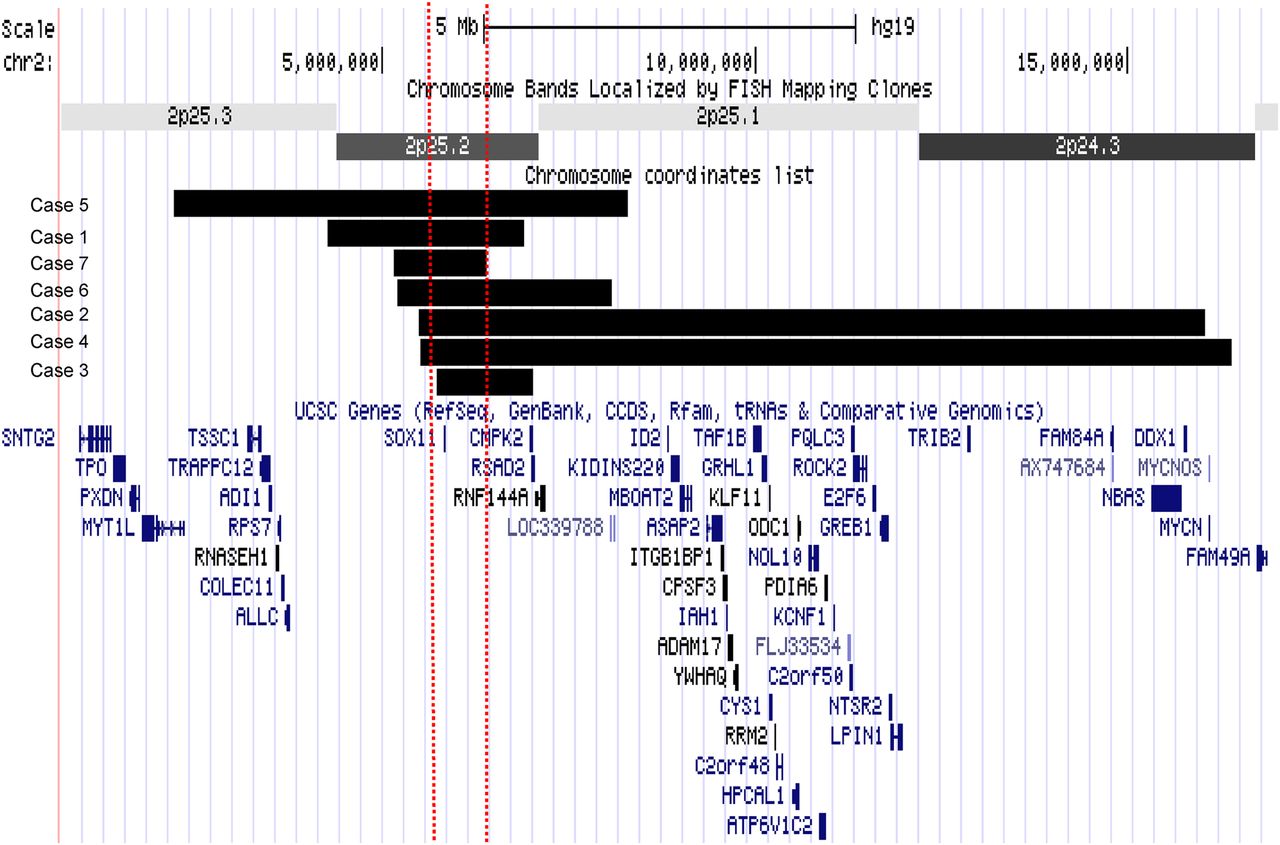
Schematic diagram of deletions. The deletions are displayed as a custom track in UCSC genome browser with refseq genes using hg 19. The horizontal black bars represent the deletion in each patient and the two dashed vertical lines represent the smallest region of overlap. The smallest region of overlap contains only the SOX11 gene.

Clinical photographs of study participants. (A). Case 4: facial photograph in top panel, photograph of hands in lower panel (note fifth finger clinodactyly). (B). Case 2: facial photograph in top panel, photograph of hands in lower panel (note fifth finger clinodactyly). (C) Case 3: facial photograph in top panel, photograph of hands in lower panel (note fifth finger clinodactyly). (D) Case 1: facial photograph in top panel. Reproduced from Lo-Castro et al.17 (E) Boy with c.150G>C (p. Lys50Asn) SOX11 mutation (case 9). Facial photograph in top panel, hand in middle panel (note fifth finger clinodactyly) and foot in lowermost panel (note broad hallux and 2–3 toe syndactyly). (F) Boy with c.87C>A (p. Cys29*) SOX11 mutation (case 10). Facial photograph in top panel, hand in middle panel (note fifth finger clinodactyly) and foot in lowermost panel (note broad hallux, 2–3 toe syndactyly and hypoplasia of nail of fifth toe). (G). Girl with c.359C>A (p. Pro120His) SOX11 mutation (case 8). Facial photograph in top panel, hand in middle panel (note fifth finger clinodactyly) and foot in lowermost panel (note broad hallux and hypoplasia of nail of fifth toe). (H) Case 7. Facial photograph in top panel, photograph of hands in lower panel (note fifth finger clinodactyly and small nails on fifth finger).
Case 2 is a 5-year 11-month-old girl, the first child of healthy non-consanguineous parents. She was born at 36 weeks of gestation by caesarean section because of intrauterine growth restriction. Birth weight was 1850 g (<10th centile) and head circumference 31 cm (<10th centile). She first walked independently at 22 months old and spoke her first words at 18 months. At 5 years and 11 months old, weight was 14.5 kg (<3rd centile), height 102.5 cm (<3rd centile) and OFC 47 cm (<2nd centile). She had troublesome gastro-oesophageal reflux and a high degree hypermetropia. On examination she had sparse hair, epicanthic folds and a wide mouth with thick lips. Bilateral clinodactyly of the fourth and fifth fingers with generalised joint laxity was noted. Brain MRI showed agenesis of the posterior third of the corpus callosum. CGH was reported as arr 2p25(5511851–16027633)x1(hg 19). The deletion encompassed the SOX11 and MYCN genes. The deletion was not present in either parent.
Case 3 is a 14-year-old girl, the first child of healthy non-consanguineous parents. She was born at 37 weeks of gestation. Birth weight was 2840 g (9th centile) and head circumference 33 cm (2nd centile). She had a weak suck and difficulty feeding in the neonatal period. She first walked at the age of 20 months. Her fine motor skills were impaired. She had no speech delay. At 14 years old, her height was 158 cm (50th) and head circumference was 52 cm (<2nd). She was dysmorphic with a high forehead, narrow palpebral fissures, a smooth philtrum, micrognathia and a high arched palate. A scoliosis, inverted nipples, bilateral fifth finger clinodactyly and 2–3 toe syndactyly were noted. CGH was reported as arr 2p25(5838893–7023548)(hg 19)x1. The deletion included SOX11, RSAD2 and CMPK2 genes. The deletion was not found in the mother. The father was not available for testing.
Case 4 is a 25-year-old woman, the third child of healthy non-consanguineous parents. Pregnancy was complicated by reduced fetal movements. Birth weight was 3130 g (25th centile). Choanal stenosis and persistent ductus arteriosus were present in the neonatal period. She first walked at 4 years and spoke at 3 years old. At the age of 25 years old, her height was 162 cm (25th centile), weight 50 kg (10th centile) and head circumference 49 cm (<0.4th centile). She has severe intellectual disability and aggressive behaviour. She is physically well but has hypermetropia. She has a flat face with upslanting palpebral fissures and wide mouth with thick lips. Short second fingers with bilateral fifth finger clinodactyly was present along with skin syndactyly of toes 2–3 and 4–5. CGH was reported as arr 2p25(5535091–16398225)(hg 19)x1 with deletion of SOX11 and MYCN. The deletion was not found in either parent.
Case 5 is a 34-year-old man, the third child of healthy non-consanguineous parents (patient 16 from the study of De Rocker et al18). He was born at term. Birth weight was 3750 g (75th). He had intellectual disability, speech delay and epilepsy. On examination at age 34 years, height was 150 cm (3rd centile) and head circumference 53 cm (3rd centile, corrected for height). He had facial dysmorphism: microretrognathia, short philtrum and mild trigonocephaly. Scoliosis and cryptorchidism were also noted. CGH was reported as arr 2p25(2231163–8300707)(hg 19)x1 with deletion of the SOX11 and MYT1L genes. This was paternally inherited; a detailed phenotypic description of the father was not available, but he was reported to be epileptic and had intellectual disability and speech delay.18
Case 6 is a 13-year-old girl, ascertained via DECIPHER with limited information. She was born at term. She presented as an infant with feeding difficulties, microcephaly and seizures. CGH was reported as arr2p25(5209876–8078809)(hg 19)x1 with deletion of SOX11, RSAD2, CMPK2 and RNF144A. It was not possible to test parental samples.
Case 7 is a 2-year-old girl, the first child of healthy non-consanguineous parents. She was born at 39 weeks of gestation by elective caesarean section. Birth weight was 3.5 kg (50th). There were feeding difficulties and poor weight gain in the first month of life. This resulted in hospital admission. Gastro-oesophageal reflux was diagnosed and the feeding difficulties resolved with gaviscon. At this time generalised hypotonia was noted. There was a history of mild gross motor delay; she first sat independently at 10 months and walked at 15 months. At the age of 2 years, she had begun to run. There were no concerns relating to fine motor skills. There was mild speech delay; at the age of 2 years, she was not producing two word phrases. She was physically well. On examination her head circumference was 47 cm (50th centile) and weight 10 kg (2nd centile). She had deep set eyes, full cheeks and thick lips. There was sparse scalp hair. She had bilateral fifth finger clinodactyly and hypoplasia of the nails of the fifth fingers and toes. CGH was reported as arr2p25.2(5166592–6400126)(hg19)x1. Only the SOX11 gene was deleted, and it was not possible to test parental samples.
Individuals with SOX11 mutations
Case 8 is a 12-year and 6-month-old girl, the first child of healthy non-consanguineous parents. She was born at 41 weeks of gestation. Birth weight was 3118 g (26th centile). There was poor feeding in the neonatal period requiring nasogastric feeding. She had global developmental delay; first walking at 2 years and 6 months old. She has never spoken. At the age of 12 years and 6 months, her height was 89.4 cm (<0.4th), weight 12.15 kg (<0.4th) and head circumference 46.5 cm (<0.4th). She has profound intellectual disability, severe autism, absence seizures, bruxism and a tendency to pick at her skin. On examination facial dysmorphism (sunken eyes, depressed nasomaxillary area and wide mouth with thick lips), bilateral fifth finger clinodactyly and hypoplasia of the toe nails of her little toes were noted. She had oculomotor apraxia. Brain MRI was normal. CGH was normal. Exome sequencing identified a de novo, heterozygous missense variant in SOX11 (c.359C>A, p. Pro120His; NM_003108.3).
Case 9 is an 11-year-old boy, the first child of healthy non-consanguineous parents. Pregnancy was complicated by third trimester bleeding. He was born at 40 weeks of gestation. Birth weight was 3500 g (50th). He was readmitted to hospital at 3 weeks of age with poor feeding (immature suck, choking and regurgitation of bottle feeds) and failure to thrive. He had global developmental delay; first walking at age 2 years and speaking his first words at age 3. At 11 years of age, he was described as being aggressive with poor attention and no sense of danger. He spoke only in short sentences and was in a special needs school. On examination he was on the 9th centile for height and weight and had a head circumference under the 0.4th centile. He had bilateral conductive deafness, cryptorchidism, hypermetropia and squint. He had malar flattening, a short philtrum and tented upper lip and a left preauricular skin tag. Bilateral 2–3 cutaneous toe syndactyly and fifth finger clinodactyly was also noted. CGH was normal. Exome sequencing identified a de novo, missense variant in SOX11 (c.150G>C, p. Lys50Asn).
Case 10 is a 6-year-old boy, the child of healthy non-consanguineous parents. He was born by planned caesarean section with a birth weight of 4.08 kg (75th). He presented with mild global developmental delay; first walking at 16 months of age and having significantly delayed expressive speech. He attends a learning unit and has over-excitable behaviour. He is generally well apart from enuresis. He was 124.5 cm tall (75th centile), 29 kg (91st centile) and head circumference was 54 cm (75th centile). On examination he was noted to have slightly coarse facial features with wide mouth and thick lips, a prominent metopic suture, arched well-defined eyebrows and midline anterior hairline upsweep. He had oculomotor apraxia. Bilateral fifth finger clinodactyly and fifth fingernail hypoplasia was noted along with bilateral fourth/fifth toenail hypoplasia. Radiographs of the hands and feet were normal. A brain MRI detected hypoplasia of the inferior aspect of the cerebellar vermis. Array CGH (180k, Agilent) was normal. Exome sequencing identified a de novo, nonsense variant in SOX11 (c.87C>A, p. Cys29*).
In silico analysis of SOX11 missense variants
The c.150G>C (p. Lys50Asn) variant was predicted by SIFT to be deleterious (score of 0) and PolyPhen to be probably damaging (score of 1). The c.359C>A (p. Pro120His) variant was predicted by SIFT to be deleterious (score of 0) and by PolyPhen to be probably damaging (score of 0.996). Both mutations localise to the HMG (DNA binding) domain (figure 3). The ‘Have Your Protein Explained’ tool identified that in the c.359C>A (p. Pro120His) variant, the mutant residue (histidine) is larger and more hydrophilic than the WT amino acid. This was predicted to interfere with DNA binding and protein–protein interaction. In the c.150G>C (p. Lys50Asn) variant, asparagine is noted to be of smaller size and neutral charge compared with the WT amino acid. This was also predicted to interfere with DNA binding. Both variants are found at evolutionary conserved amino acids. Neither variant was found in the exAC database, nor was the c.87C>A, p. Cys29* mutation.

SOX11 variants identified in the current study. (A) Schematic diagram of SOX11 protein demonstrating location of three reported sequence variants. The p.S60P and p.Y116C variants reported by Tsurusaki et al8 are also shown, the structural effects of these mutations can be found in reference 8. (B and D) Models demonstrating alteration of SOX11 protein structure associated with the two missense variants. Green areas represent the wildtype residue while the red area indicates the structure adopted by the mutant amino acid. Both the missense variants were in the DNA binding domain of SOX11 and predicted to alter its structure, thus interfering with DNA binding. (C) Bar chart demonstrating that the twoSOX11 missense variants had reduce ability to activate the GDF5 promoter in an in vitro reporter system. The adjacent western blot confirms that the mutant proteins were stably expressed during the experiment.
In vitro assessment of effect of SOX11 mutations on transcriptional activity
Luciferase reporter assays in HeLa cells indicate that both the p.Lys50Asn and p.Pro120His variants display reduced ability to activate the GDF5 promoter compared with WT protein (figure 3).
Knockdown of Sox11 in xenopus laevis
Knockdown of Sox11 by MO injection resulted in a significant reduction in head area and interpupillary distance compared with controls (both p<0.0001 on Mann–Whitney U test). Figure 4 shows a representative morphant and control. There was no increased death rate among the morphants and the controls.

Sox11 knockdown leads to microcephaly in Xenopus laevis. (A) Bilateral injection of Sox11 MO results in significant smaller heads measured by the head area (white dotted circles) and the pupillary distance (red lines) compared with bilateral control MO injections. In addition, Sox11 morphants show an eye phenotype as previously described (red arrowhead; Cizelsky et al14). (B) Statistical evaluation of the measured head area. (C) Statistical evaluation of the measured pupillary distance. N, number of individual embryos analysed. ****, p≤0.0001. p Values were calculated by a non-parametric Mann–Whitney rank sum test.
Expression of SOX11 in human brain
For all brain regions examined, SOX11 expression levels (RNA-seq) were highest in the first trimester of pregnancy and then fell significantly (Kruskal–Wallis, p<0.01) to reach a nadir in the fourth decade of life (figure 5A–D). SOX11 expression levels (microarray) were significantly greater in brain areas with high levels of neurogenesis compared with areas of low neurogenesis (Mann–Whitney U test p<0.01) (figure 5E) at both 15 weeks and 21 weeks of gestation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of SOX11 in developing human brain. (A–D) Changes in SOX11 expression levels as measured by RNA-sequencing in the cerebellum, hippocampus, prefrontal cortex and striatum, respectively. Columns labelled first, second and third refer to trimesters of pregnancy. Column 10 represents the first decade of life, 20 the second decade of life and 40 the third and fourth decades. There was a significant decline in SOX11 expression levels with increasing age as assessed by the Kruskal–Wallis test (p<0.01). (E) Microarray data demonstrating that SOX11 is expression is higher in brain regions with high levels of neurogenesis (periventricular) compared with areas with low levels of neurogenesis (cerebellum, thalamus, brain stem), *p<0.01. The first two columns represent 15 weeks of gestation while the second two columns represent 21 weeks of gestation.
Discussion
Here we report seven patients with chromosome 2p25 deletions, which include SOX11, and three individuals with de novo SOX11 point mutations. The individuals with deletions had several phenotypic features in common (table 1). Microcephaly was reported in all but two. All had developmental delay, with particularly marked speech delay. A shared facial dysmorphology of wide mouth and thick lips could be discerned. One of the patients reported here had trigonocephaly, and so it is interesting to note that a 2004 report describes a boy with trigonocephaly, cleft palate and multiple minor anomalies with deletion of SOX11 resulting from an unbalanced translocation 46,XY,t(2;17)(p25;q24).19 Fifth finger clinodactyly and cutaneous syndactyly of toes 2–3 was a frequent examination finding. There was considerable variability between deletion cases—likely reflecting the different CNV sizes. The individuals with de novo SOX11 mutations had several shared phenotypic features. Microcephaly, low birth weight and neonatal feeding difficulties associated with hypotonia were frequent findings. Developmental delay was reported in all cases apart from patient 2 in Tsurusaki et al's8 report. Hypoplasia of the distal phalanx of the fifth finger, broad halluces, 2–3 toe syndactyly and hypoplasia of the nail of the fifth toe are also frequent. Ocular abnormalities were present in both deletion and mutation cases; case 1 had right microphthalmia, cases 2, 4 and 7 had hypermetropia and squint while cases 7 and 9 had oculomotor apraxia. Our report confirms that SOX11 mutations and deletions can be associated with a neurodevelopmental disorder, which manifests features of CSS.
The clinical features associated with SOX11 deletion or mutation overlap with other syndromes. Hypoplasia of the fifth fingers with dysmorphism and intellectual disability can be observed in mosaic trisomy 9,20 deafness-onychodystrophy–osteodystrophy–mental retardation syndrome21 and phenytoin embryopathy.22 There is overlap between mild forms of Cornelia de Lange syndrome and individuals with SOX11 mutations.23 It is important to note that none of our cohort had a clinical diagnosis of CSS prior to CGH or exome sequencing being performed. In retrospect, the individuals we report had clinical features compatible with CSS (eg, hypoplasia of fifth finger) but did not present with classical dysmorphic features of CSS, which would enable a clinical diagnosis to be readily made. This is analogous to the presentation of ARID1B mutations. ARID1B mutations have been identified as the most common cause of CSS and also in children with intellectual disability who have subtle features of CSS, but who would not have been diagnosed with this syndrome on the basis of their phenotype alone.24 However, we suggest that certain features of CSS, such as the pattern of hair distribution (synophrys, sparse scalp hair but increased body hair) and fifth finger hypoplasia in combination, should lead to CSS being included in differential diagnoses for children with a neurodevelopmental disorder.
Several other genes within the deleted 2p25 regions could contribute to the observed phenotypes. In the centromeric deletions (cases 2 and 4), it is highly likely that MYCN deletion contributes to the phenotype. Deletions and mutations of MYCN are associated with Feingold syndrome.25 The classical features of Feingold syndrome are microcephaly, intestinal atresias and brachymesophalangy of the second and fifth fingers. The severe intellectual disability reported in case 4 is unusual for individuals with Feingold syndrome.25 In the individual with a telomeric deletion (case 5), loss of MYTL1 is likely to contribute to the phenotype. However, case 5 has borderline microcephaly, while the other individuals in the report of De Rocker et al18 who had similar deletions involving MYT1L but not SOX11 tended to have macrocephaly. This suggests that SOX11 haploinsufficiency may exert a powerful, negative influence on brain growth. Cases 1 and 7 have no genes other than SOX11 in the deleted region, while the deletion in cases 3 and 6 also contains the CMPK2, RSAD2 and RNF144A genes. CMPK2 encodes a mitochondrial nucleoside monophosphate kinase,26 RSAD2 encodes viperin, which is an antiviral protein,27 and RNF144A is an E3 ubiquitin ligase involved in DNA damage repair and apotosis.28 Haploinsufficiency scores indicate that heterozygous loss of these genes is unlikely to cause a neurodevelopmental disorder. This provides evidence that SOX11 deletion alone can be associated with a neurodevelopmental phenotype.
The fact that both heterozygous deletions and mutations of SOX11 are associated with microcephaly suggests that loss of function and haploinsufficiency may be the underlying mechanism. The two missense variants we describe are within the HMG DNA binding domain (as were the two previously reported missense variants8) while the c.87 C>A variant would be predicted to lead to premature termination of translation prior to this domain. SOX11 is a single exon gene. Nonsense-mediated decay (NMD) may not occur with mutations in the final exon of a gene,29 so it is possible that a mutant SOX11 transcript may not undergo NMD. However, the c.87C>A mutation occurs before the HMG domain in SOX11, so it is likely that any protein product will be unable to bind DNA and induce gene expression. The mutant protein is also likely to be unstable given its truncated nature. It is thus likely that all three mutations will interfere with the ability of SOX11 to regulate its target genes. Our luciferase reporter gene assays provide further evidence in support of this as they indicate a reduced ability of mutant SOX11 to induce gene expression. An in vitro study of SOX11 overexpressing cells identified multiple genes upregulated by SOX11, which are relevant to neurogenesis and brain development.10 We hypothesise that haploinsufficiency of SOX11 could potentially reduce expression of these target genes at critical points in brain development, resulting in a neurodevelopmental disorder.
The expression pattern of SOX11 in the human brain is in keeping with the gene playing a role in neurogenesis during embryonic development. We show that SOX11 expression peaks in the first three months of in utero life and declines thereafter. Since neurogenesis in the fetal brain is largely completed by mid-gestation,2 this temporal expression pattern fits with SOX11 being involved in neurogenesis in the fetal brain. The spatial expression pattern of SOX11 in fetal brain also suggests that SOX11 is involved in neurogenesis since SOX11 expression was significantly higher in the ventricular zone than in areas with relatively low levels of neurogenesis. That two of our cases had brain malformations provides further evidence that SOX11 functions in human neurodevelopment.
Data from animal models also suggests that SOX11 plays an important role in eye development since SOX11 knockdown in zebrafish can cause ocular malformations.4 In addition, variants in SOX11 have also been identified in two individuals with iris coloboma and no neurodevelopmental phenotype.4 This may be explained by the fact that the sequence variants in these individuals were not located in the DNA binding HMG domain of SOX11 protein, while variants reported herein associated with CSS were predicted to interfere with DNA binding.
Our experiments in Xenopus embryos indicate that loss of Sox11 is associated with microcephaly. We previously demonstrated that Sox11 depletion leads to smaller eyes.14 This is in line with our current study as case 1 shows microphthalmia. Moreover, the microophthalmia phenotype can be rescued by co-injection of WT Sox11 RNA.14 The fact that (1) Sox11 depletion leads to an eye phenotype14 similar to case 1 (our current study), (2) co-injection of Sox11 MO together with WT Sox11 RNA results in a rescue of the Sox11 MO-induced eye phenotype14 and (3) Sox11 MO injection does not lead to an increased death rate compared with control MO injection suggests that the microcephaly phenotype of Sox11 knockdown in X. laevis is not explained by non-specific toxic effects of MO injection. The precise mechanism by which loss of Sox11 results in microcephaly in Xenopus, however, is still unclear. Our previous work on Xenopus eye development indicates that Sox11 knockdown does not alter proliferation but is associated with increased neuronal apoptosis.14 This suggests that SOX11 may also function as a neuronal survival factor in brain development.
In conclusion, we describe a series of individuals with SOX11 deletions or de novo mutations presenting a neurodevelopmental disorder, which had clinical features compatible with CSS. The two SOX11 missense variants reported here are the only plausibly pathogenic SOX11 variants identified from over 1000 exomes performed on probands with a developmental disorder in the DDD study.30 SOX11 variants are a rare cause of neurodevelopmental disorders. That both deletions and mutations of SOX11 can cause CSS is in keeping with data reported for ARID1A and ARID1B, deletions or truncating mutations of which also cause CSS.31 Deletion mapping, in our small cohort, suggests that SOX11 deletion, although likely pathogenic in itself, can also act as part of a contiguous gene deletion along with loss of MYT1L or MYCN. The mechanism is likely to be SOX11 haploinsufficiency with dysregulation of the SOX11 target genes and consequent disruption of brain development. SOX11 is itself induced by the BAF (BRG1-associated or HRBM-associated factors) complex, leading to neuronal differentiation.32 It is noteworthy that many other genes mutated in CSS function in the BAF complex.32 The majority of genes in the BAF complex in which mutations have been identified are associated with a CSS phenotype (eg, ARID1A, ARID1B, SMARCB1, SMARCA4 and SMARCE1), while mutations in SMARCA2 cause Nicolaides–Baraitser syndrome.31 Our report thus reinforces the importance of the BAF complex in CSS. In conclusion, the current study provides evidence that SOX11 is a further member of the SOX protein family associated with human neurodevelopmental disease.
Acknowledgments
We thank the families who kindly agreed to be part of this study. This study makes use of data generated by the DECIPHER Consortium. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. We thank Dr Samantha Knight, Oxford University, for performing the initial microarrays in case 9.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Correction notice This article has been corrected since it published Online First. The 7th author's name has been amended.
Contributors AH: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (performed and analysed xenopus experiments). AP: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (SOX11 sequencing and validation). MB, SM and VM: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (clinical characterisation of study participants). —. —. IK, IS, YT and NM: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (SOX11 reporter studies). — —. —. AL-C, GP, BA and AB: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (clinical characterisation of participants). — — — JT, MH and DK: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (exome studies, securing funding). — — SK: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (Xenopus studies). UK: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (exome studies, securing funding, clinical characterisation). AS: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (clinical characterisation.). AM: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work (conceptualisation of study, clinical characterisation, wrote first draft of manuscript). In addition, all of the authors drafted the work or revising it critically for important intellectual content; and gave final approval of the version to be published; agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding Funding for the project was provided by the Wellcome Trust. We declare that those who collected data and deposited it in the DECIPHER database bear no responsibility for its use and interpretation in the current work. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme. We also thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and Medical Research Council Hub grant G0900747 91070) for generating the sequencing data for case 10.
Disclaimer The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health.
Competing interests AM is supported by the INSIGNEO collaboration for in silico medicine at Sheffield University.
Ethics approval The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC).
Provenance and peer review Not commissioned; externally peer reviewed.