Article Text

Abstract
CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated nuclease 9) systems have emerged as versatile and convenient (epi)genome editing tools and have become an important player in medical genetic research. CRISPR-Cas9 and its variants such as catalytically inactivated Cas9 (dead Cas9, dCas9) and scaffold-incorporating single guide sgRNA (scRNA) have been applied in various genomic screen studies. CRISPR screens enable high-throughput interrogation of gene functions in health and diseases. Compared with conventional RNAi screens, CRISPR screens incur less off-target effects and are more versatile in that they can be used in multiple formats such as knockout, knockdown and activation screens, and can target coding and non-coding regions throughout the genome. This powerful screen platform holds the potential of revolutionising functional genomic studies in the near future. Herein, we introduce the mechanisms of (epi)genome editing mediated by CRISPR-Cas9 and its variants, introduce the procedures and applications of CRISPR screen in functional genomics, compare it with conventional screen tools and at last discuss current challenges and opportunities and propose future directions.
- Genetics
- Genetic screening/counselling
- Genome-wide
Statistics from Altmetric.com
Introduction: limitations of conventional genetic screen approaches
Genetic screens aim at identifying the causal relationship between genotype and phenotype through introducing perturbations into genome and/or epigenome so as to investigate how (epi)genome aberrations result in phenotype alterations. Conventionally, genetic screens in cultured cells are mainly conducted with the aid of RNA interference (RNAi) or complementary DNA (cDNA) libraries.1 ,2 However, RNAi can only partially suppress gene expression and thus its application is limited to knockdown screens.1 ,3 Moreover, due to the endogenous nature of RNAi pathway, it often incurs pervasive off-target events because of extensive endogenous interactions.4 These pervasive off-target effects may confound the interpretation of screen results. cDNA library-mediated gain-of-function screen, on the other hand, also has some limitations. First, it is impossible for a cDNA library to cover the full complexity of the transcriptome in a cell. And second, the expression of cDNAs is not subjected to endogenous regulation and thus is often expressed at a non-physical or aberrantly high level. Recently, the emergence of CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated nuclease 9) techniques5–7 offers a novel and versatile platform for genetic screen studies. In this review, we introduce the mechanisms and merits of CRISPR-Cas9 and its variants in genome and epigenome editing, retrospect their applications in genetic screens studies, discuss current challenges and propose future directions.
Mechanisms of (epi)genome editing by CRISPR-Cas9 and its variants
Wild type (wt) Cas9 for genome editing
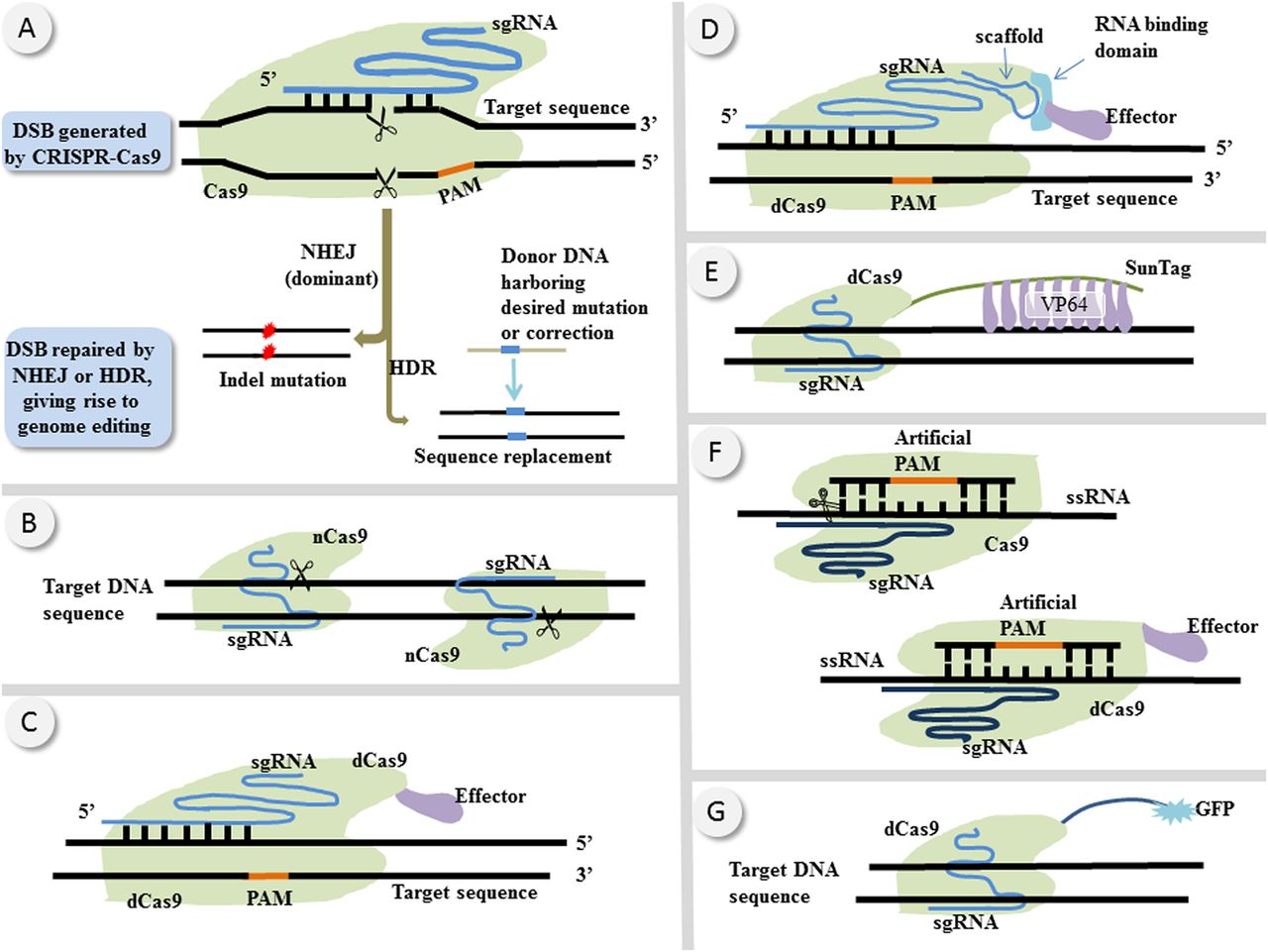
CRISPR-Cas9 is a kind of programmable nuclease derived from microbial immune system.5–7 Its nuclease Cas9 is directed by a single guide RNA (sgRNA) that is complementary with the target DNA strand. Upon the presence of a short sequence termed protospacer-adjacent motif (PAM) on the opposite DNA strand, sgRNA binds to the target strand by complementarity and thus guides Cas9 to generate site-specific double-strand breaks (DSBs) on the target DNA sequence (figure 1A).6–8 The resultant DSBs are subsequently processed by DNA repair mechanisms such as non-homologous end joining (NHEJ) or homologous directed repair (HDR).6 ,7 In general, NHEJ is more active than HDR because the latter requires a homologous template and is mainly restricted to S and G2 phases of the cell cycle.9 HDR can result in precise gene replacement whereas NHEJ is error prone in that it may induce frameshift indel mutations that may abrogate target gene function10 ,11 (figure 1A).

Schematic of (epi)genome editing by CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated nuclease 9) and its variants. (A) With the presence of a PAM sequence on the opposite DNA strand, sgRNA binds to the target strand by base pairing and directs the nuclease Cas9 to generate site-specific DSBs on the target DNA sequence. The DSB is then repaired either by NHEJ or by HDR. The former may cause frameshift indel mutations that may abolish the target gene function whereas the latter may give rise to precise gene replacement. (B) Cas9 has two catalytic domains, the inactivation of either of which produces a nickase Cas9 (nCas9) that generates single-strand break instead of DSBs. A pair of nCas9s can produce paired nicks which were reported to incur less off-target effects compared with wild type Cas9 that generates DSBs.13 ,14 (C) If the two catalytic domains of Cas9 are inactivated, wild type Cas9 is turned into catalytically dead Cas9 (dCas9) that when fused to epigenetic modifiers can modulate target gene expression. (D) A scaffold RNA (scRNA) capable of recruiting RNA-binding proteins (RBPs) can be incorporated into sgRNA. These RBPs are then tethered to epigenetic modifiers and exert site-specific epigenetic regulations.19 ,20 (E) A protein scaffold termed SunTag that is capable of recruiting up to 10 copies of VP64 is fused to dCas9 to increase the potency of transcriptional regulation.16 ,43 (F) Upon the existence of an artificial PAM sequence, Cas9 is reprogrammed into RNA-targeting (RCas9) that can bind and cut single-stranded RNA (ssRNA) site-specifically.62 Catalytically dead RCas9 (dRCas9)62 can serve as a site-specific ssRNA binding domain fused to various effectors, which holds potentials to exert RNA modulations. (G) Fluorescent protein fused to dCas9 is directed by sgRNAs to enable living cell imaging of genomic loci of interest.63 sgRNA: single guide RNA; PAM, protospacer-adjacent motif; DSB, double-strand breaks; NHEJ, non-homologous end-joining; HDR, homologous directed repair; GFP: green fluorescent protein.
CRISPR-Cas9 is a RNA-guided nuclease and therefore possesses several advantages in genome editing over conventional protein-guided programmable nuclease such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). First, in CRISPR-Cas9 technology, multiple genomic loci can be targeted simultaneously using multiple complementary sgRNAs.6–8 Second, retargeting of ZFN and TALEN requires laborious processes of redesign and resynthesis of guide proteins, whereas retargeting of CRISPR-Cas9 needs only the synthesis of a complementary sgRNA sequence, which is much easier and simpler.
CRISPR-Cas9 variants for (epi)genome editing
Besides wt-CRISPR-Cas9, CRISPR-Cas9 system has several variants that have distinct properties and functions in genome and epigenome editing. Nickase Cas9 (nCas9), which is produced by inactivating either of the two catalytic domains (called HNH and RuvC, respectively) of wt-Cas9, generates single-strand break instead of DSBs on the target DNA sequence12 ,13 (figure 1B). It was reported that a pair of nCas9 producing paired nicks incur much less off-target cleavage compared with wt-Cas9 that generate DSBs, possibly for the reason that the off-target single nicks are quickly repaired and thus undetectable.13 ,14
If the two catalytic domains of Cas9 are inactivated, wt-Cas9 is turned into catalytically dead Cas9 (dCas9) that when fused to epigenetic modifiers such as VP6415 ,16 or KRAB16 ,17 can mediate activation or suppression of target gene expression, respectively (figure 1C). However, the magnitude of regulatory effects of dCas9-modifier on target gene expression is generally low17–19 and varies greatly across different sgRNAs.17 More robust expression regulation can be achieved with multiple sgRNAs targeting a single gene.15 However, in genome-wide screen studies, robust gene regulation with individual or only a few sgRNAs is preferred over multiple sgRNAs strategy because the size of sgRNA library cannot be unlimitedly expanded. To increase the regulatory potency with individual sgRNA, a scaffold RNA (scRNA) capable of recruiting RNA-binding proteins (RBPs) can be incorporated into sgRNA.19 ,20 These RBPs are then tethered to epigenetic modifiers to exert site-specific epigenetic regulations (figure 1D) on target genes.19 ,20 Moreover, the modifiers tethered to scRNA and the ones fused directly to dCas9 have synergic effects on target gene regulation.19 ,20 This scRNA-dCas9 strategy has already been applied to genome scale activation screens recently.19
Strategies and procedures of genetic screens with CRISPR-Cas9
There are two foundations for the application of CRISPR-Cas9 technology in screen studies: the availability of oligonucleotide synthesis technology21 that enables mass synthesis of sgRNA library; and the multiplexing nature of CRISPR-Cas9 system that enables multiple genomic editing simultaneously. CRISPR screen combines the programmability of RNAi with the versatility of CRISPR-Cas9 techniques in genome and epigenome editing. Using CRISPR-Cas9 and its variants, researchers can carry out multiple forms of screen studies such as wt-Cas9-mediated loss-of-function (LOF) screens22–24 and dCas9-mediated activation16 ,19 or knockdown screens.16
There are generally two screen formats: arrayed screen and pooled screen. In arrayed screen, individual or a small pool of reagents are prepared separately and arrayed in multiwell plates.25 ,26 Because the genetic perturbation in each well is already known in advance in arrayed screen, it enables a wide range of cellular phenotypes to be observed and investigated. In the pooled screen format, on the contrary, sgRNA or RNAi library is massively synthesised, cloned and delivered into cells to introduce various genetic perturbations.22–24 ,27 ,28 After cells with intended phenotype are physically separated, their genetic perturbations are read out to propose a causal link between the phenotype and genotype.22–24 ,27 ,28 Pooled screen is less expensive and less laborious than arrayed screen, and it can be used in in vivo studies.24 ,28 However, pooled screen format is limited to phenotypes such as cell proliferation and survival or phenotypes that are selectable using cell sorting techniques.
Currently, CRISPR screen is mainly carried out in pooled format (figure 2), in which sgRNA library is massively synthesised and delivered into the initial cell population. After genetic perturbation, cells with intended phenotype must be physically separated from the remaining population. There are two ways to select cells with intended phenotype: positive selection and negative selection. Positive selection aims at identifying genetic perturbations that enable cells to survive and/or proliferate under a certain selective pressure, such as the one that is toxic or intolerable to the initial cell population.22–24 Therefore, positive selection is especially suitable for identifying mutations that confer resistance to a certain adverse environment, such as drug treatment, pathogen infection or hypoxia. Because there are generally only a few perturbations capable of inducing protective mutations and cells with such protective perturbations continue to proliferate, these protective perturbations are usually greatly enriched in the final cell population and thus easy to be detected. Negative selection, on the contrary, aims at identifying genetic perturbations that cause cells to be depleted from the population over time, such as perturbations that abolish the function of genes essential for cell survival or proliferation.29–31 These perturbations can be ferreted out by comparing sgRNA presentations at the beginning and at the end of screen period. In pooled screen format, negative selection requires more sensitive screen readout methods and more efficient sgRNAs for several reasons. First, the magnitude of sgRNA depletion in negative selection is more modest than that of sgRNA enrichment in positive selection. Second, the number of depleted perturbations in negative selection is usually bigger than that of enriched perturbations in positive selection.

{kind=link}
{kind=link}
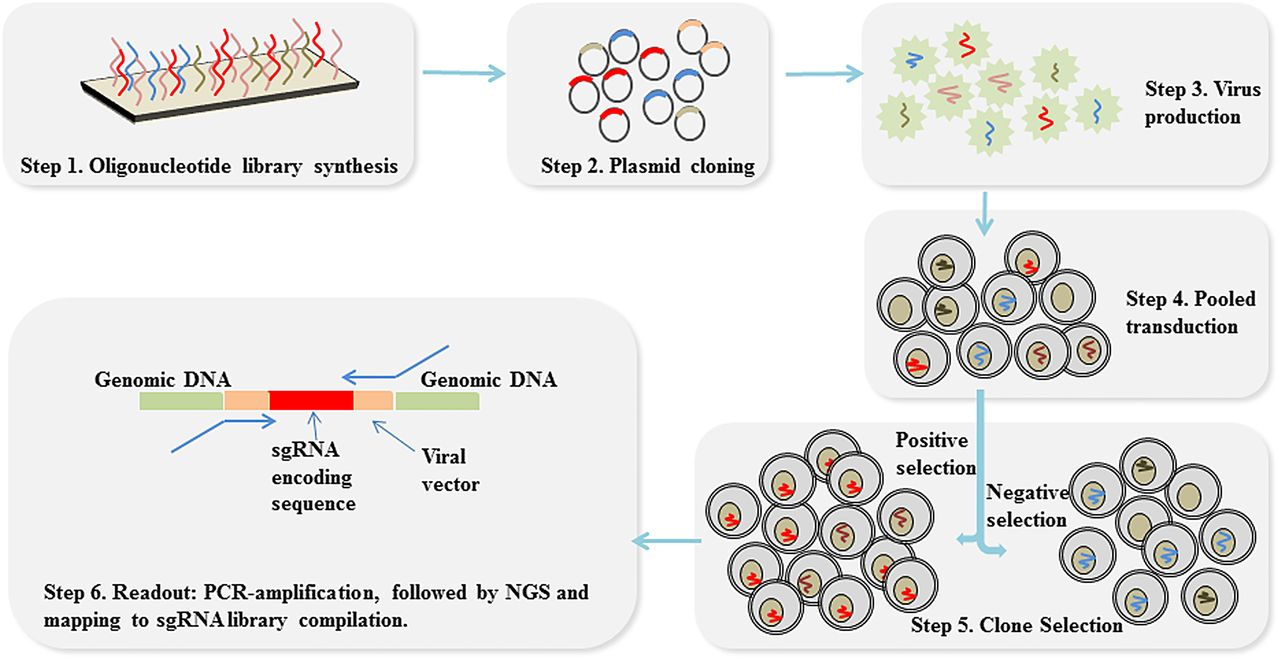
Flow chart of CRISPR (clustered regularly interspaced short palindromic repeats) screen in pooled format. sgRNA library is massively synthesised (step 1), cloned into plasmids (step 2) and packaged into viral vectors (step 3). Cas9-expressing cells are infected with these vectors at low multiplicity of infection in order that most cells receive only one virus integration (step 4). After positive or negative selection (step 5), sgRNA-encoding regions are PCR-amplification and sequenced, followed by NGS and mapping to the sgRNA library design to read out sgRNA representation in the selected cell population (step 6). sgRNA: single-guide RNA; NGS, next generation sequencing.
After positive or negative selection, genomic DNA is extracted from the selected cell subpopulation and the sgRNA-encoding regions are subjected to PCR amplification. Subsequently, these regions are sequenced and mapped to the pre-existing sgRNA library to read out sgRNA representation in the selected cell subpopulation16 ,19 ,22 (figure 2). By comparing the sgRNA profiles during different screen periods (eg, at the beginning and at the end of screen), sgRNA enrichment or depletion can be identified in order to draw a causal link between genetic perturbation and the observed phenotype.
Applications of CRISPR genetic screens
LOF screen
As mentioned above, Cas9-generated DSBs in the target sequence are dominantly repaired by NHEJ. NHEJ may result in frameshift indel mutations that may abolish target gene function, which is the basis for CRISPR-Cas9-mediated LOF screens. CRISPR-Cas9-mediated LOF screen can be used in vitro and in vivo for multiple purposes.
Identification of genes involved in resistance to drugs, toxins or infectious diseases
The first examples of Cas9-mediated LOF screen are two contemporaneous studies by Wang and colleagues22 and Shalem and colleagues23 Wang and colleagues22 developed a genome-scale lentiviral sgRNA library and then transfected it into Cas9-expressing KBM7 cells (a chronic myelogenous leukaemia cell line) to identify genes whose LOF mutations confer resistance to 6-thioguanine (6-TG). 6-TG is lethal to wt-KBM7 cells but not to cells with DNA mismatch repair (MMR) pathway deficiency. As a result, sgRNAs targeting one of the four critical genes in MMR pathway were remarkably enriched in the final cell population, with all of the top 20 hits targeting one of the four genes, suggesting a low frequency of off-target editing. The authors then conducted another genome-scale CRISPR screen in haematological tumour cell lines22 and identified TOP2A or CDK6 as genes whose LOF may confer resistance to etoposide, a chemotherapeutic agent. Interestingly, each of the 20 sgRNAs targeting TOP2A or CDK6 was remarkably enriched in the final cell population, suggesting a high reagent consistency.
In the study by Shalem and colleagues,23 the authors transduced a library of 64 751 sgRNAs targeting 18 080 genes into melanoma cells to identify LOF mutations that render resistance to vemurafenib (an antimelanoma agent).23 These mutations are potential targets for developing resistance-reversing therapeutics. Subsequent validation experiment with individual sgRNAs showed a high validation rate (the majority of the top-hit genes showed a phenotypical effect when knocked out individually) and high reagent consistency (the majority of the independent sgRNAs targeting the same gene showed a consistent phenotypical effect). The reagent consistency of this CRISPR screen23 was much higher than that of a previous RNAi screen study32 on vemurafenib resistance. In another genome-scale CRISPR screen study,33 27 known and 4 novel candidate genes were identified whose LOF mutation may confer resistance to either Clostridium septicum α-toxin or 6-TG in mouse embryonic stem cells. Similarly, in a subsequent validation process, the percentage of top-hit sgRNAs that can individually generate the desired phenotypical effect was significantly higher than that of shRNAs, indicating a high reagent consistency.
Apart from identifying mutations that confer resistance to drugs or toxins,22 ,23 ,32–34 CRISPR screen can also be used for searching for host gene mutations that may provide protection against infectious diseases. West Nile virus (WNV) infection can cause neuroinvasive disease featured by massive neuronal cell death, the mechanism of which remains unclear. In 2012, there were 5674 WNV cases reported in the USA.35 ,36 Ma and colleagues37 conducted a genome-wide CRISPR screen with a library comprising 77 406 sgRNAs followed by a second subpool screen for conformation. The authors identified seven genes with the strongest phenotypical effects whose LOF mutations conferred protection against WNV-induced cell death, albeit with no effect on WNV replication. These genes are promising targets for developing preventive strategies or treatment methods for WNV infectious diseases.
Interrogation of gene functions in health and diseases
Interrogation of gene functions in physiological or pathological states is another application of Cas9-mediated LOF screens. Parnas and colleagues,38 for example, transduced genome-wide sgRNA libraries into dendritic cells and sorted these cells based on the expression levels of tumour necrosis factor (TNF) with a fluorescent antibody by flow cytometry. By comparing the sgRNA profiles between cells with high and low TNF levels, genes that regulate the expression of TNF in response to bacterial lipopolysaccharide were identified, including previously known ones and novel candidates, many of which were subsequently validated with individual sgRNAs.
The study by Chen and colleagues24 serves as another example of CRISPR screen-meditated gene function interrogation. The authors transduced a genome-wide lentiviral sgRNA library together with Cas9-expressing lentiviral vectors into a non-metastatic mouse lung cancer cell line. In a xenograft model, infected cells were found to form primary tumours more quickly than controls and metastasised into the lungs, which is beyond the ability of the original cell line, implying that some of the gene mutations accelerated tumour growth and conferred metastatic potential. Subsequent deep sequencing revealed a high sgRNA dropout rate during tumour evolution, with less than half of the sgRNAs retained in the early tumours, less than 4% in late tumours and less than 0.4% in the metastases. This is in line with the clonal selection processes during tumour evolution. The genes targeted by the ‘top-ranked’ sgRNAs in late-stage tumours and metastases are candidates responsible for enhanced malignancy, among which are well known oncosuppressors and novel candidates. Subsequently, the malignancy-promoting potentials of some of the top hits were validated by individual sgRNAs or sgRNA mini pools, showing a high validation rate.24 This study presented a practical method for high-throughput identification of driver mutations underlying tumour evolution and metastasis, which can be readily adopted for functional genomic studies of other cancer types.
Compared with conventional knockdown screen with RNAi, CRISPR-Cas9-mediated LOF screen possess several advantages. First, CRISPR screen induces irreversible alterations into the DNA sequences whereas RNAi acts on transcripts by degrading RNA or inhibiting translation, which are incomplete and subjected to various epigenetic interactions. In cases when the phenotypical effects of LOF mutation rather than gene knockdown are the interest, CRISPR screen is more suitable than RNAi screen. Second, the effects of RNAi screen are limited to transcribed loci where CRISPR screen can cover many intranscribed regions, such as enhancers, promoters and intergenic regions. Moreover, existing evidence has shown that CRISPR-Cas9-mediated LOF screen has high validation rate22–24 and high reagent consistency,22 ,23 lending support to its reliability. However, given the limited number of evidences available up to date, its reliability remains to be tested. As for off-target effects, although minimal or tolerable22 ,23 off-target effects have been reported in LOF CRISPR screen, these results were all based on sequencing of potential off-target regions and hence may underestimate the real profile of off-target effects. Unbiased methods such as chromatin immunoprecipitation sequencing,39 ,40 GUIDE-seq41 and Digenome-seq42 are still needed to provide a more comprehensive evaluation of off-target effects.
Activation and knockdown screens
As stated above, apart from wt-CRISPR-Cas9 that can introduce permanent alterations into the genome, CRISPR-Cas9 has several variants that can be applied to epigenome regulation or transcriptome modulation (figure 1B–F), thus holding potentials in transcriptional modulation screens (TMSs). The genome and epigenome are two sides of one coin, they interact with each other and cooperatively function in health and diseases. dCas9-mediated TMS and Cas9-mediated LOF screen can be used complementarily to provide a comprehensive profile of functional genome and epigenome.
Weissman JS and colleagues, for example, achieved gene knockdown (CRISPR interference, CRISPRi) by fusing KRAB (a transcriptional suppressor) to dCas9 and achieved gene activation (CRISPR activation, CRISPRa) by fusing to dCas9 a protein scaffold termed SunTag (figure 1E), which is capable of recruiting up to 10 copies of VP64.16 ,43 These dCas9-effectors were directed by sgRNAs to exert locus-specific transcriptional regulations. Using sgRNA libraries coupled with these dCas9-effectors, the authors16 conducted genome-wide activation and knockdown screens in positive selection and negative selection formats, and identified genes essential for cell survival as well as genes modulating sensitivity to toxins.16 Previous studies have shown that for CRISPR-Cas9, off-target binding is more pervasive than off-target cleavage,44–46 raising concern that dCas9-mediated TMSs might incur pervasive off-target effects. However, this study16 demonstrated that the CRISPRi activity of sgRNA/dCas9-KRAB is highly sensitive to mismatches between sgRNAs and the target sequences, indicating that the observed off-target binding in previous studies44–46 might be too transient to have off-target transcriptional effects. Moreover, in this study,16 up to 99.7% of negative sgRNA controls had no detectable activity, lending support to the high specificity of this method.
In a study conducted by Konermann and colleagues,19 gene activation in a melanoma cell line that rendered resistance to vemurafenib, an antimelanoma agent, were identified via dCas9/scRNA-mediated activation screen. In this activation screen,19 transcriptional activators such as p65 and HSF1 were recruited and directed to the target gene by scRNAs (figure 1D) to exert site-specific transcriptional activation. Because only cells with mutations conferring resistance to vemurafenib can survive and proliferate, sgRNAs enriched in the final cell population were candidates that were responsible for the acquired vemurafenib resistance.19 The method presented in this study19 can be readily adapted to screening for activation mutations that sustain tumour growth and progression under special circumstances such as hypoxia, radiation exposure or chemotherapy, thus providing potential antitumour strategies in these contexts.
Conventionally, activation screens are mainly carried out with cDNA library. However, as stated above, gene expression from cDNA library is ectopic and non-physical. dCas9-mediated activation screens, on the contrary, act on and cause expression upregulation from endogenous genomic loci. Nonetheless, to what extent this expression upregulation can preserve endogenous regulation and feedback remains to be investigated in the future. As for off-target activity, existing evidence has suggested that dCas9-mediated TMSs might incur only minimal off-target effects, as evidenced by minimal transcriptional modulation activity of non-targeting sgRNAs,16 ,19 undetectable off-target activity of individual sgRNAs,19 and sensitivity of CRISPRi activity to mismatches between sgRNAs and their targets.16 However, these are all indirect evidences that are still not concrete enough to support a final conclusion. Genome-wide unbiased methods39–42 remain needed to provide a more direct and reliable profile of off-target effects.
Perspectives and concluding remarks
From the above examples, we can see that CRISPR screen requires efficient delivery methods, high on-target efficiency and low off-target effects. There are several ways to increase the efficiency of delivering CRISPR components into cells. Viral vectors are currently the most commonly used vectors in CRISPR screen. They can be engineered to increase their tropism for cells of interest and thus to improve delivery efficiency.47 ,48 Among various viral vectors available, adeno-associated virus (AAV) vectors are the most commonly used ones owing to their low immunogenicity, broad tissue tropism and minimal insertional mutagenesis.49 However, the size of the commonly used Cas9 from Streptococcus pyogenes (SpCas9) renders it challenging to be transduced with AAV vectors due to the limited cargo of the latter. Reassuringly, Cas9 has several orthologues that are derived from other bacteria such as Staphylococcus aureus50 ,51 and Streptococcus thermophiles.51 Because these orthologues are smaller in size than SpCas9 and can be easily transduced with AAV vectors, they hold application potentials in CRISPR screen studies in the future to improve delivery efficiencies. Moreover, the delivery of purified recombinant Cas9 protein rather than Cas9 nucleotide was reported to enhance editing efficiency significantly.14 Rational design of sgRNAs and careful selection of PAM sequences can also improve on-target efficiency16 ,51–54 because a library of sgRNAs with strong target-binding potency and high genome coverage is of vital importance for the success of CRISPR screen. As stated above, the target range of commonly used SpCas9 is restrained by the NGG PAM requirement, which is a problem in genome-wide screen studies. A recent study51 demonstrated that the target range of Cas9 can be broadened by engineering SpCas9 to recognise alternative PAM or by using Cas9 orthologues from other bacteria with novel PAM specificities. As for strategies to reduce off-target effects, they have been extensively reviewed elsewhere,55–58 such as double-nicking by a pair of Cas9 nickases13 (figure 1B), sgRNA truncation59 and the fusion of dCas9 with FokI nuclease,60 ,61 which might be adopted in the future to increase CRSIPR screen specificity. A recent study by Vidigal and colleagues,62 for example, reported a one-step method to clone sgRNA pairs into vectors, which can generate paired sgRNAs library that together with paired nCas9 holds application potentials in functional genomic screens.
Given the rapid advance in CRISPR-Cas9 techniques, we can envisage several future directions of CRISPR screen. First, RNA-targeting Cas9 (RCas9) is a kind of Cas9 variant that can site-specifically bind and cut single-stranded RNA (ssRNA)63 upon the presence of an artificial PAM sequence (figure 1F). RCas9 can be further reprogrammed into catalytically dead RCas9 (dRCas9)63 with functions mirroring those of dCas9. dRCas9 can serve as a site-specific ssRNA binding domain fused to various effectors to exert modulations on RNA sequences (figure 1F). RCas9 and dRCas9 might be harnessed in the future to conduct transcriptome screen to identify transcriptome aberrations that confer phenotypical features such as growth advantage, metastasis ability or drug resistance. Second, fluorescent proteins fused to dCas9 can be directed by sgRNAs and thus enable living cell imaging of genomic loci of interest64 (figure 1G), which can be used in CRISPR screens to enable direct and quantitative assessment of mutational dynamics. Third, apart from genome-scale sgRNA libraries, CRISPR screens can also be conducted in more flexible manners, that is, sgRNA mini libraries or subpools that focus on specific pathways or particular biological processes. This focused library strategy has already been harnessed in several studies65 ,66 and is especially suitable for knowledge-based screenings. Fourth, inducible Cas967–70 that can introduce genetic disruptions in a phase-specific and/or cell-specific manner can be applied to CRISPR screen to study the phage-specific effects of mutations or the phenotypical effects of mutations in different orders. Combining inducible Cas9 and sgRNA subpools together, researchers may be able to investigate the casual relationship and/or competitive dynamics of different signalling pathways in physiological or pathological conditions.
In conclusion, CRISPR screen provides a practical and high-throughput way for functional genomic studies. Compared with conventional screens conducted with RNAi or cDNA libraries, CRISPR screen is more versatile because multiple screen formats can be carried out with the aid of wt-CRISPR-Cas9 system and its variants, such as LOF screen, knockdown screen and activation screen. Moreover, existing evidence suggests that CRISPR screen is generally more reliable and more specific than RNAi screen.16 ,19 ,22–24 ,32 ,33 The versatility, reliability and specificity of CRISPR screen render it a promising player in medical genetic researches. CRISPR screens have been applied to various functional genomic studies such as identifying genes essential for cell survival, genes involved in resistance to drugs or toxins and genes promoting cancer cell metastasis. Future improvements in sgRNA/Cas9 design, synthesis, selection and delivery will help improve the specificity and efficiency of CRISPR screen. Potential applications of CRISPR screen in medical research include areas such as transcriptome screen, CRISPR screen in combination with living genomic imaging, and inducible Cas9 that enables spatiotemporal specific screen. Given the rapid advance in this area, we can optimistically envisage that in the near future CRISPR screen might transform functional genomic researches.
References
Footnotes
Hui-Ying Xue, Li-Juan Ji and Ai-Mei Gao are co-first authors.
Contributors X-JL and J-DH conceived the idea and wrote the majority of the manuscript. H-YX and L-JJ searched and read through literatures, A-MG and PL wrote part of the manuscript. All authors have read and approved the manuscript.
Funding This work was supported by the Project Fund of Health Bureau of Jiangsu Province (No. H201358), Yangfan Project of Shanghai Science and Technology Commission (14YF1411900) and National Natural Science Foundation of China (81402949).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement This is a review article rather than an original research article. So, data sharing statement is inapplicable.