Article Text

Abstract
Background Congenital hyperinsulinism (HI) can have monogenic or syndromic causes. Although HI has long been recognised to be common in children with Beckwith–Wiedemann syndrome (BWS), the underlying mechanism is not known.
Methods We characterised the clinical features of children with both HI and BWS/11p overgrowth spectrum, evaluated the contribution of KATP channel mutations to the molecular pathogenesis of their HI and assessed molecular pathogenesis associated with features of BWS.
Results We identified 28 children with HI and BWS/11p overgrowth from 1997 to 2014. Mosaic paternal uniparental isodisomy for chromosome 11p (pUPD11p) was noted in 26/28 cases. Most were refractory to diazoxide treatment and half required subtotal pancreatectomies. Patients displayed a wide range of clinical features from classical BWS to only mild hemihypertrophy (11p overgrowth spectrum). Four of the cases had a paternally transmitted KATP mutation and had a much more severe HI course than patients with pUPD11p alone.
Conclusions We found that patients with pUPD11p-associated HI have a persistent and severe HI phenotype compared with transient hypoglycaemia of BWS/11p overgrowth patients caused by other aetiologies. Testing for pUPD11p should be considered in all patients with persistent congenital HI, especially for those without an identified HI gene mutation.
- Endocrinology
- Epigenetics
- Genetics
Statistics from Altmetric.com
Introduction
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycaemia in infancy. In 65% of cases, it is associated with inactivating mutations in either ABCC8 or KCNJ11,1 and in a small number of cases, it has been associated with Beckwith–Wiedemann syndrome (BWS).2–5 Notably, ABCC8 and KCNJ11 are located on chromosome 11p15.1, proximally adjacent to the BWS/11p overgrowth spectrum (11p overgrowth) imprinted regions (figure 1). These genes encode the sulfonylurea receptor 1 (SUR-1) and inwardly rectifying potassium (Kir6.2) subunits of the plasma membrane ATP-sensitive potassium (KATP) channel in pancreatic beta cells.6–9 Inactivation of KATP channel activity leads to persistent membrane depolarisation, elevated cytosolic calcium and insulin release, even in the context of low-plasma glucose levels. A focal form of congenital HI is caused by paternal transmission of a monoallelic loss-of-function mutation of one of the two KATP subunits, compounded with an embryonic pancreas-limited chromosome parental recombination of 11p15 to result in a pancreatic lesion with uniparental isodisomy for chromosome 11p (pUPD11p) and biallelic loss of function of the KATP channel.10 Children with focal KATP-HI are unresponsive to treatment with diazoxide, a KATP channel agonist, but can be cured by surgical excision of the small focal lesion.1 Of note, despite a proven paternal parent-of-origin effect, the KCNJ11 and ABCC8 genes do not have imprinted gene expression. This raises a key question regarding the physiological mechanism underlying HI in these patients and suggests other imprinted elements on 11p15.

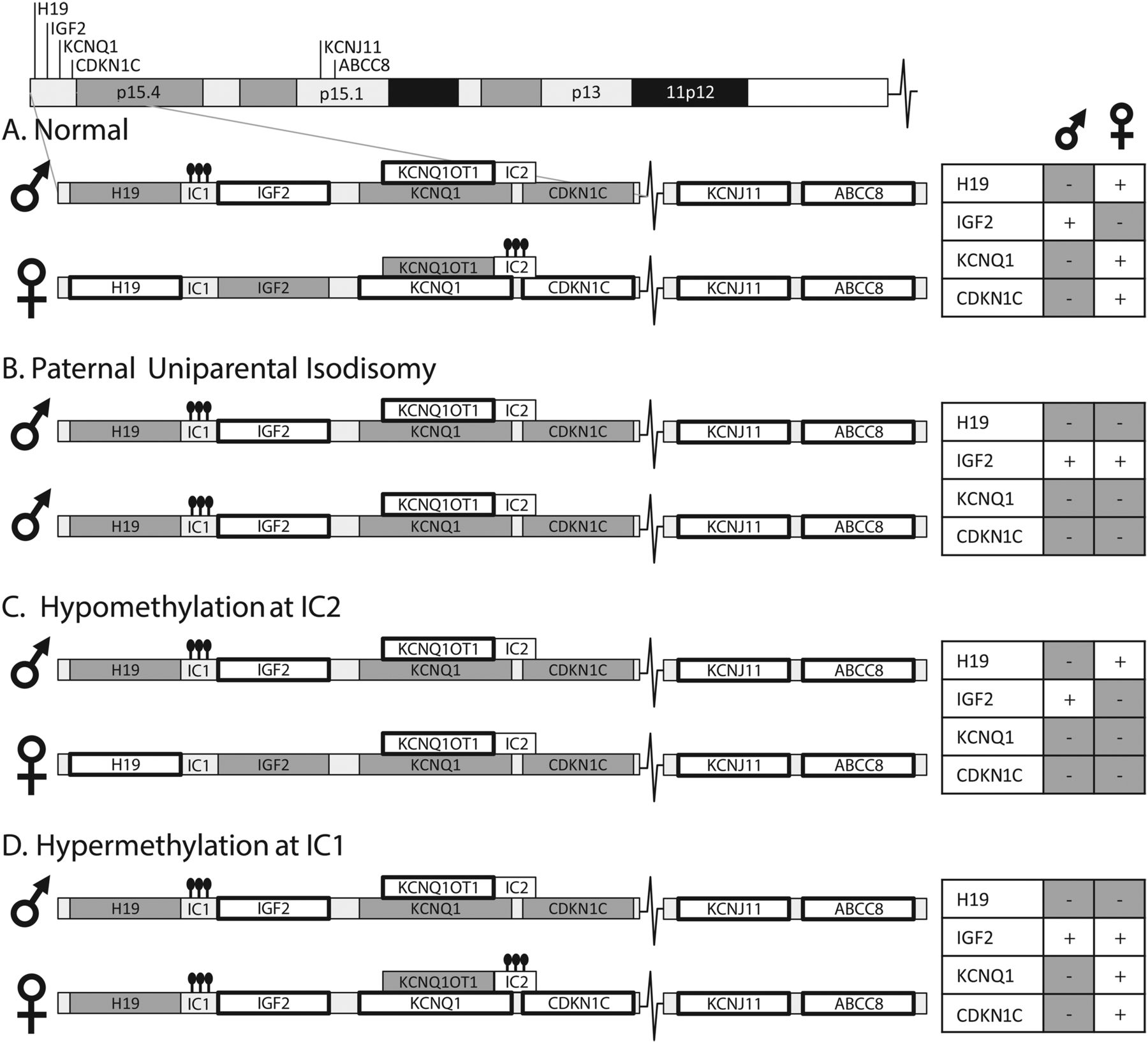
Mechanisms of Beckwith–Wiedemann syndrome (BWS). Chromosome 11 includes the BWS region and the KATP genes. Expressed genes are noted with a bold box surrounding. The hyperinsulinism (HI) genes KCNJ11 and ABCC8 are biallelically expressed. (A) 11p15 contains two adjacent imprinting control regions that are altered in BWS. Normally, imprinting control region 1 (IC1) is methylated on the paternal allele resulting in paternal expression of IGF2 and unmethylated on the maternal allele resulting in maternal expression of H19. Imprinting control region 2 (IC2) is methylated on the maternal allele resulting in KCNQ1 and CDKN1C (p57) expression and unmethylated on the paternal allele. Closed circles indicate methylation. (B) In paternal uniparental isodisomy (pUPD11p), IC1 is methylated on both alleles resulting in biallelic IGF2 expression and loss of H19 expression. IC2 is unmethylated on both alleles, resulting in loss of KCNQ1 and CDKN1C expression. (C) Hypomethylation at IC2 leads to loss of KCNQ1 and CDKN1C expression. (D) Hypermethylation at IC1, a rarer cause of BWS, leads to biallelic IGF2 expression and loss of H19 expression. Maternally transmitted CDKN1C mutations, and rare deletions or duplications of the imprinted region(s) can also lead to BWS.
Genetic or epigenetic changes on chromosome 11p15.5 are well known to lead to BWS. BWS is an overgrowth disorder whose classical features include macrosomia, macroglossia, hemihypertrophy, distinct facial features, including ear and infraorbital creases, and embryonal tumours.11 The 11p overgrowth spectrum connotes some, but not all, features of BWS.12 Hypoglycaemia occurs in approximately 50% of classic BWS cases. The hypoglycaemia can be mild and typically resolves within the first few days of life.13–15 However, 5% of classic BWS cases have persistent hypoglycaemia caused by HI that requires medical and/or surgical management. The mechanism of HI in patients with BWS and its relationship to specific aetiologies of BWS has remained unexplained for decades.
The genetic or epigenetic changes causing BWS typically occur during embryonal development in a subset of cells, leading to a mosaic mixture of normal and BWS cells and resulting in a mosaic spectrum of clinical features.16 The BWS locus on 11p15.5 includes two adjacent regions of imprinted genes, including IGF2, a growth-promoting protein, and two growth-inhibitory genes; the long non-coding RNA H19 and the cell cycle regulator p57/CDKN1C (see figure 1). Methylation at two key sites plays an important role in regulating gene expression of these loci (see figure 1). Approximately 50% of BWS is caused by isolated hypomethylation of the imprinting control region 2 (IC2) on 11p15, leading to loss of expression of both p57/CDKN1C and of KCNQ1, a voltage-gated potassium channel, as shown in figure 1. Another 5–10% of BWS is due to isolated hypermethylation at the imprinting control region 1 (IC1), leading to loss of H19 and biallelic expression of IGF2 (figure 1).11 Finally, 20% of BWS cases are caused by paternal uniparental isodisomy for chromosome 11p (pUPD11p), which leads to a combined loss of p57/CDKN1C and KCNQ1 expression as well as biallelic IGF2 expression (figure 1). This same genetic change, pUPD11p, can lead to classic BWS, but can often lead to more subtle features including isolated hemihypertrophy of a limb or isolated overgrowth of an organ.16 ,17 The entire group of disorders caused by genetic or epigenetic alterations on chromosome 11 can be referred to as ‘11p overgrowth spectrum’ or ‘11p overgrowth’. This spectrum, therefore, includes patients with classic BWS clinical features and also includes patients with subtle hemihypertrophy and with HI.12
Based on our experience in several patients with congenital, severe, persistent HI and BWS/11p overgrowth associated with pUPD11,4 we hypothesised that HI in 11p overgrowth could be due to pUPD11 that either uncovers a paternally transmitted KATP mutation or initiates a KATP-independent mechanism. The purpose of this study was to examine a large cohort of 11p overgrowth cases with HI in order to define the relationship between the mechanism of 11p overgrowth, the presence or absence of KATP mutations and the phenotype of HI. The results in 28 patients suggest that persistent HI may be uniquely associated with the pUPD11p mechanism of 11p overgrowth.
Methods
Patient cohort
We reviewed all children with HI who were referred to the Children's Hospital of Philadelphia (CHOP) Hyperinsulinism Center between 1997 and 2014 for those with features of BWS. The diagnosis of HI was based on previously described criteria.18 ,19 Severe HI was defined as congenital, persistent hypoglycaemia that requires medical therapy with diazoxide, octreotide or continuous feedings or surgical intervention. Patients were defined to be responsive to diazoxide if hypoglycaemia was well controlled on a dose ≤15 mg/kg/day as demonstrated by either maintaining plasma glucose >70 mg/dL for 12–18 h of fasting or developing hyperketonemia (beta-hydroxybutyrate >2 mmol/L) before plasma glucose fell to 50 mg/dL. Octreotide responsiveness used the same criteria but with a subcutaneous octreotide dose between 5 and 15 µg/kg/day. The diagnosis of 11p overgrowth was based on clinical features (table 1). However, in several cases, pancreatic histology was the only feature suggestive of 11p overgrowth.
Beckwith–Wiedemann syndrome/11p overgrowth spectrum clinical features
Mutation analysis
Genomic DNA was isolated from peripheral blood (5 PRIME, Gaithersburg, Maryland, USA) or from saliva (Oragene DNA self-collection kit; DNA Genotek, Kanata, Ontario, Canada). DNA from surgical pancreatic specimens or cultured from skin fibroblasts was extracted using the DNA/RNA Allprep kit (QIAGEN, Valencia, California, USA). Mutation analysis for ABCC8, KCNJ11, GCK, UCP2 and MEN1 was performed as previously described.1
Single-nucleotide polymorphism array
Genome-wide single-nucleotide polymorphism (SNP) array analysis was performed as previously described.20 This method is capable of detecting mosaicism as low as 3% for paternal UPD of the 11p region.16
Methylation analysis
Methylation patterns at IC1 and IC2 on chromosome 11p15 were analysed using allele-specific methylated multiplex real-time quantitative PCR.21
Histology
Histology studies were performed on pancreatic tissue obtained at the time of surgery. Sections were stained with primary mouse monoclonal antibodies directed against p57Kip2/CDKN1C as a marker for paternal UPD11p (ThermoScientific, Clone 57P06, 1:300), insulin (ThermoScientific, INS04+INS05, 1:400), and chromogranin.
Results
Patients
Between 1997 and 2014, a total of 702 children with HI were referred to CHOP. Of the 501 without focal HI, 28 children had findings of BWS/11p overgrowth (table 1). Of these 28 11p overgrowth children, 26 had pUPD11p (subsequently referred to as UPD-11p overgrowth). Further, 6 of the UPD-11p overgrowth patients were diazoxide-responsive, 6 were treated with octreotide and 14 required surgery. The two cases with HI and BWS/11p overgrowth not due to pUPD11p were associated with hypomethylation at IC2 (patients 27 and 28); these two had only mild HI: patient 27 was responsive to low doses of diazoxide and patient 28 had only transient HI lasting 3 weeks.
As shown in table 1, the most common features of 11p overgrowth were hemihypertrophy (24/28), macrosomia (22/28), hepatomegaly (20/28) and umbilical hernia (17/28). Almost half of the patients who required surgery had two or fewer major diagnostic features of 11p overgrowth. Hemihypertrophy was present in 24 of 28 of these patients and, in several cases, was the only feature of 11p overgrowth. There was a significant correlation between per cent mosaic pUPD11p in blood and the number of clinical BWS features (p<0.0009), but no correlation between BWS clinical features and per cent pUPD11p in pancreas.
As shown in table 2, the extent of pancreatectomy in the most UPD-11p overgrowth patients who required surgery ranged from 60% to 100%. One patient had biopsies only and was managed with continuous dextrose through a gastrostomy; another patient had a 2% resection. Glucose infusion rates (GIRs) in the 14 patients prior to surgery ranged from 5 to 50 mg/kg/min (median 20 mg/kg/min). The four patients with both pUPD11p and KATP mutations (#2, 3, 7 and 9, table 3) had higher glucose requirements (25–50 mg/kg/min) and more severe clinical courses. One of the four patients who died prior to surgery had a paternally transmitted KATP mutation. Only five of the surgical cases did not require additional hypoglycaemia management after surgery.
Hyperinsulinism clinical features and management
Hyperinsulinism gene mutations identified
By 2 years 17 out of 20 surviving patients were off of all treatment; only two patients required treatment beyond 2 years of age (table 2).
Clinical course of illustrative UPD-11p overgrowth HI patients
Two cases illustrate the range of HI and 11p overgrowth clinical features in our cohort.
Patient 6 had clinical features of BWS, pUPD11p in multiple tissues and no identified KATP mutations. He was born at 37 weeks gestation with a birth weight of 4.04 kg (+3.1 SD) with multiple features of BWS (macrosomia, macroglossia, hemihypertrophy, bilateral ear creases and organomegaly). He had severe hypoglycaemia requiring a maximum GIR of 13 mg/kg/min. He was not responsive to diazoxide or octreotide. An 18F-DOPA PET scan demonstrated a large area of increased uptake in the pancreas, and he underwent a 60% pancreatectomy. Postoperatively, he required management with nasogastric dextrose until 5 months of age when a fasting test demonstrated complete resolution of HI. SNP array and methylation testing showed variable degrees of mosaicism for pUPD11p in blood (25–30%), skin (25–30%) and pancreas (80–90%). He is currently 23 months old and undergoing regular BWS tumour screening.
Patient 2 had clear features of BWS and also carried a paternally inherited recessive KATP pathogenic variant. She had pUPD11p in blood, skin and pancreas. She was born at 38 weeks with a birth weight of 4.56 kg (+3.6 SD). Clinical features of BWS (macrosomia, macroglossia, hemihypertrophy, bilateral ear creases, umbilical hernia, hepatomegaly) and hypoglycaemia due to HI were noted at birth. Mutation analysis revealed a paternally inherited recessive pathogenic variant in ABCC8 (table 3). She was not responsive to diazoxide and required a GIR of up to 25 mg/kg/min. 18F-DOPA PET scan showed a region of increased uptake in the tail of the pancreas, and initially she underwent a 98% pancreatectomy and had a large pancreatic region of pUPD11p. Following the first surgery, she continued to require a GIR of up to 24 mg/kg/min and further resection was performed. Following the second surgery, she continued to require a GIR of 16 mg/kg/min and was discharged on octreotide and continuous nasogastric dextrose with a GIR of 9.4 mg/kg/min. Two weeks later, she developed necrotising enterocolitis, sepsis and died.
Histological findings in UPD-11p overgrowth HI
Figure 2 illustrates the range of adenomatosis seen in the UPD-11p overgrowth/HI pancreases. In general, the lesions showed a spectrum of histomorphological changes characterised by a variable, but overall increase in the volume of endocrine relative to acinar tissue. Normal lobular architecture was retained. In contrast to lesions of focal congenital HI, which are usually <1 cm in diameter, the expansion of endocrine tissue in UPD-11p overgrowth was not confined to a small localised area of proliferation, but was present throughout large areas of pancreas. Frequently, endocrine tissue within affected areas had a prominent, trabecular architectural arrangement demonstrated by a patterning of nuclei in a ribbon or garland-like pattern. Because of this distinct appearance and the large size of the lesion, pUPD11p was considered in some patients based on pancreatic histology and was confirmed by SNP arrays showing pUPD11p. Tissue sections demonstrated a gradient in the degree of islet expansion across the entire pancreas (figure 2D). The increase in islet cell expansion was accompanied by a corresponding decrease in nuclear p57 expression (figure 2E).
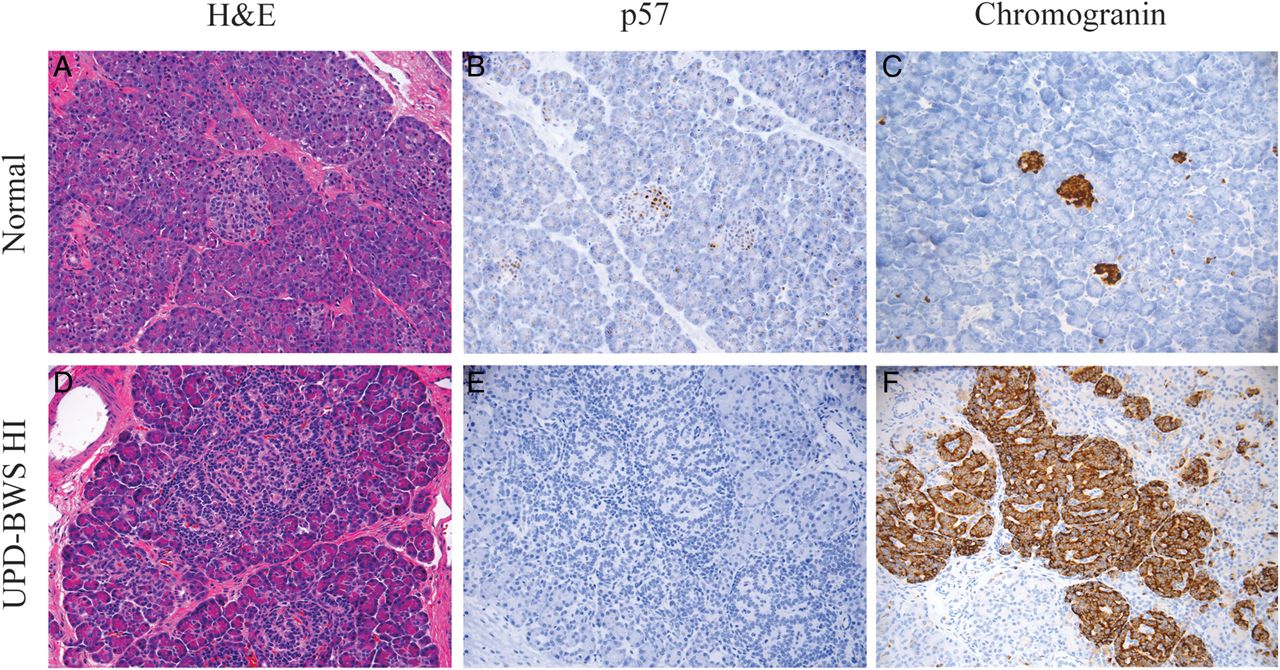
Pancreatic histology in Beckwith–Wiedemann syndrome (BWS) hyperinsulinism (HI). The histology of the BWS pancreas is similar to focal pancreatic lesions but demonstrates a larger area of affected pancreas compared with focal lesions. (A) Normal pancreas by H&E staining, (B) p57 and (C) chromogranin. (D–F) Images from a UPD-BWS patient's pancreas that demonstrate an increase in the size of islets associated with a more prominent trabecular arrangement of islet cell nuclei (D). The expanded islets and overgrown endocrine tissue correlate with a decrease in p57 (CDKN1C) expression (E). The chromogranin staining demonstrates that the region of expanded endocrine cells also includes exocrine acinar tissue (F). The variable degree of overgrown endocrine tissue with expanded islets and preservation of lobular architecture is distinct from both focal and diffuse HI histology.
Molecular analysis
As shown in figure 3, 25 of the 26 UPD-11p overgrowth patients had molecularly confirmed pUPD11p in blood, skin and/or pancreas. Methylation studies were performed simultaneously and confirmed pUPD11p in these cases. The case without detectable pUPD11p had clinical features of 11p overgrowth, but SNP array and methylation analysis from blood was normal and no pancreatic tissue was available for testing. In the absence of evidence of other causes of 11p overgrowth (ie, methylation was normal), this patient was presumed to have mosaic pUPD11p in the pancreas.

{kind=link}
{kind=link}
{kind=link}
Region of mosaic paternal uniparental isodisomy for chromosome 11p in cases of Beckwith–Wiedemann syndrome (BWS) and hyperinsulinism (HI). The location of the BWS region genes and the HI genes on chromosome 11 are indicated on the chromosome at the top of the figure. The grey bars indicate the extent of paternal uniparental isodisomy along chromosome 11 as determined by single-nucleotide polymorphism array analysis. * indicates the presence of a KATP gene mutation (see table 3). ND, not determined because a sample was not available.
The level of pUPD11p mosaicism detected in blood ranged from 0% to 98% (figure 3). In samples of lesion from resected pancreas, the per cent mosaicism ranged from 30% to 99%. In all cases, the per cent mosaicism in the affected regions of pancreas was higher compared with blood or skin.
Neither the extent of paternal UPD of chromosome 11 nor the per cent of mosaicism within various tissues (blood, skin or pancreas) correlated significantly with the severity of HI (p>0.05). For the 25 cases with documented pUPD11p, the length of the UPD segments ranged from as small as 3.5 Mb, including only the telomeric end of chromosome 11p containing the BWS/11p overgrowth gene region (2.1 Mb), to all of chromosome 11 (134.5 Mb). When samples were available from different tissues (blood, skin and/or pancreas), the length of the pUPD11p region was always consistent between samples. In seven cases, the pUPD11p region did not even include the KATP gene region (11p15.1, 17.4 Mb). Three of these seven were responsive to diazoxide and four were diazoxide unresponsive, of whom one required surgery.
Discussion
The results of this study indicate that BWS/11p overgrowth accounted for a small, but clinically important and informative fraction of the cases with congenital persistent HI referred to the CHOP Hyperinsulinism Center over a 17-year period. Notably, the cause of the 11p overgrowth in nearly all of these patients (26/28) was pUPD11p and more than three-fourths of these UPD-11p overgrowth patients had prolonged and severe HI, which failed to respond to treatment with diazoxide. In contrast, severe HI was not observed in 11p overgrowth due to causes other than pUPD11p (ie, IC2 hypomethylation). This is consistent with a recent report of 19 cases of BWS due to isolated hypomethylation of the KvDMR region (IC2), which found that half of the patients did not develop HI, and of the ones that did, none required pancreatectomy.25 Together, these data are consistent that persistent severe HI in BWS/11p overgrowth is exclusively a feature of mosaic pUPD11p and is not seen in other causes of 11p overgrowth. Notably, BWS/11p overgrowth patients who had pUPD11p in conjunction with a paternally transmitted inactivating KATP mutation had the most severe hypoglycaemia. However, in contrast to one of our initial hypotheses, most cases of HI in pUPD-11p overgrowth, including most of the severe cases requiring pancreatectomy, were not associated with a KATP mutation.
Large cohort studies of BWS report hypoglycaemia in all BWS molecular subtypes but do not distinguish between transient neonatal hypoglycaemia and severe persistent HI.26–28 Congenital, severe, persistent HI requiring surgery has been previously reported in only a very small number of cases of BWS in association with pUPD11p.2–5 A case report in 2005 suggested that the mechanism of HI was loss of KATP channel function due to abnormal localisation of the SUR-1 subunit of the KATP channel, although there was no evidence of any mutations in KATP channel subunits.2 In contrast, our results indicate that, although HI in UPD-11p overgrowth can be extremely severe and persistent when combined with a paternally transmitted KATP mutation, most cases (24 of 28) did not have an identifiable KATP defect.
As reviewed by Arnoux et al,29 it remains unknown whether the mechanism of HI in BWS/11p overgrowth is an increased mass of beta cells, a dysregulation of insulin secretion, or a combination of the two. Since most of our cases have no evidence of a KATP channel mutation, our results suggest that the mechanism of HI in UPD-11p overgrowth must involve a KATP gene-independent mechanism. Since HI appears to be specific to isodisomy for the paternal BWS imprinted genes shown in figure 1, it is possible that the HI is related to a combination of islet overgrowth due to overexpression of IGF2 and underexpression of H19 and CDKN1C, together with a lack of expression of the known maternally imprinted KCNQ1 voltage-gated potassium channel (figure 1). This channel is one of many voltage-gated potassium channels expressed in the beta cell involved in repolarisation of the plasma membrane after KATP closure. Loss of KCNQ1 expression may impair repolarisation of the beta cell and lead to failure to turn off insulin secretion. In support of this explanation, adults with long-QT syndrome due to heterozygous inactivating mutations of KCNQ1 have been reported to have HI due to exaggerated insulin responses to oral glucose.30 Although KCNQ1 expression is biallelic in many tissues, it was recently reported that KCNQ1 is exclusively expressed from the maternal allele in human fetal islets but subsequently becomes biallelic in adult islets.31 Thus, it is possible that UPD-11p overgrowth islets in early infancy have dysregulated insulin secretion due to deficient expression of KCNQ1.
A notable feature of UPD-11p overgrowth in our series was heterogeneity in the severity of HI, which was unrelated to either the degree of mosaicism in non-pancreatic tissues or the chromosomal size of the region of isodisomy. It is attractive to speculate that severity may depend on the size of the adenomatosis lesion in the pancreas, that is, that smaller lesions may be controllable by diazoxide, but larger lesions may require octreotide or even surgery to control hypoglycaemia. The use of 18F-DOPA PET scans may be helpful to assess the size of the pancreatic lesion in UPD-11p overgrowth patients and in planning the extent of pancreatic resection in patients requiring surgery.
Despite some early severity, most of the UPD-11p overgrowth patients in our series did not require treatment beyond 2 years of age. This contrasts with the monogenic forms of congenital HI, such as defects in the KATP channel, in which chronic or even lifelong treatment is usually needed well beyond 2 years.32 It is possible that the apparently earlier and more complete resolution of HI in UPD-11p overgrowth may correspond with the shift of KCNQ1 expression from fetal monoallelic imprinted expression to biallelic adult expression.31 However, further study is required to explore this mechanism of action in UPD-11p overgrowth.
The clinical recognition of UPD-11p overgrowth HI patients is complicated by the fact that physical features of BWS may not be readily apparent. In addition to cases that fulfil some BWS criteria, we have three additional cases of HI, not included in this series that fit into the 11p overgrowth spectrum. These cases had pancreatic adenomatosis, HI without a KATP defect, and minimal clinical features of BWS and illustrate the extreme subtle end of the 11p overgrowth spectrum. This is supported by the findings that pUPD11p may be present in pancreas, but not be detectable in peripheral tissues, such as blood or skin. We have noted that hemihypertrophy appears to be the most useful indication of UPD-11p overgrowth HI in our series. In some cases, an 18F-DOPA PET scan may be helpful in detecting the presence of a large islet adenomatosis lesion in the pancreas. The challenge of recognising clinically subtle 11p overgrowth features in cases subsequently shown to have pUPD11p suggests that there may be more cases of UPD-11p overgrowth HI than previously appreciated.
Despite the difficulty in noting features associated with 11p overgrowth, it is important to recognise UPD-11p overgrowth in patients with both mild or severe forms of HI since up to a quarter of BWS/11p overgrowth patients with pUPD11p are at risk of developing tumours during childhood.27 This is also true in cases with very subtle 11p overgrowth features including one case of HI being the only presenting feature of 11p overgrowth and the patient developing a hepatoblastoma.16 ,17 Due to these additional clinical implications, UPD-11p overgrowth HI should be considered in cases with large focal pancreatic lesions with or without a KATP mutation, even in the absence of overt features of 11p overgrowth. To facilitate identification of these potentially subtle patients, we emphasise a high index of suspicion, recommend sensitive SNP array and methylation analysis in patients with congenital HI, and encourage examination for clinical features of UPD-11p overgrowth by an experienced geneticist. The choice between SNP array or methylation analysis depends on the sensitivity of the available test for detecting mosaicism, and we recommend using the most sensitive test available in each instance. Additionally, because the KATP genes are on chromosome 11p, it is also important to screen for recessive mutations that may be unmasked in patients with UPD-11p overgrowth if there is persistent hypoglycaemia.
In summary, persistent HI in BWS/11p overgrowth appears to be exclusively due to pUPD11p and may present to be either diazoxide-responsive or diazoxide-unresponsive. In cases of UPD-11p overgrowth that have also have a paternally transmitted KATP mutation, HI may be particularly severe and difficult to control. Recognition of UPD-11p overgrowth in patients with congenital HI is of particular importance because of the need to monitor for potential tumours.
References
Footnotes
Contributors JMK, KEB, CAS and MAD contributed to the acquisition, analysis, and data interpretation and drafting of the manuscript. TRB, AG, LKC, SAB, SG, LM, AAP, NSA and DDDL contributed to the acquisition, analysis, data interpretation and revising the manuscript for intellectual content. All coauthors have approved the final version of the manuscript.
Funding This work was supported in part by grants from the National Institutes of Health (R37 DK056268 to CAS, KL2TR000139 and T32GM008638 to JMK, UL1RR024134 and UL1TR000003), and Alex's Lemonade Stand Foundation Young Investigator Award to JMK.
Competing interests None declared.
Ethics approval The Children's Hospital of Philadelphia IRB.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Except for the raw data, no additional data are available. Raw data may be made available if it does not infringe patients’ rights.