Article Text

Abstract
Introduction Gene panel testing for breast cancer susceptibility has become relatively cheap and accessible. However, the breast cancer risks associated with mutations in many genes included in these panels are unknown.
Methods We performed custom-designed targeted sequencing covering the coding exons of 17 known and putative breast cancer susceptibility genes in 660 non-BRCA1/2 women with familial breast cancer. Putative deleterious mutations were genotyped in relevant family members to assess co-segregation of each variant with disease. We used maximum likelihood models to estimate the breast cancer risks associated with mutations in each of the genes.
Results We found 31 putative deleterious mutations in 7 known breast cancer susceptibility genes (TP53, PALB2, ATM, CHEK2, CDH1, PTEN and STK11) in 45 cases, and 22 potential deleterious mutations in 31 cases in 8 other genes (BARD1, BRIP1, MRE11, NBN, RAD50, RAD51C, RAD51D and CDK4). The relevant variants were then genotyped in 558 family members. Assuming a constant relative risk of breast cancer across age groups, only variants in CDH1, CHEK2, PALB2 and TP53 showed evidence of a significantly increased risk of breast cancer, with some supportive evidence that mutations in ATM confer moderate risk.
Conclusions Panel testing for these breast cancer families provided additional relevant clinical information for <2% of families. We demonstrated that segregation analysis has some potential to help estimate the breast cancer risks associated with mutations in breast cancer susceptibility genes, but very large case–control sequencing studies and/or larger family-based studies will be needed to define the risks more accurately.
- Cancer: breast
Statistics from Altmetric.com
Introduction
Genetic testing of families with multiple cases of breast and/or ovarian cancer often targets the youngest affected woman (index case) in each family. Clinical genetic testing in this context has been largely limited to the BRCA1 and BRCA2 genes (unless additional indicators are present) until recently. For most women with breast cancer, these tests are uninformative as they do not identify a clearly pathogenic mutation in either gene.1 Many other putative breast cancer susceptibility genes have been identified, with varying levels of evidence for their association with breast cancer. Today, diagnostic testing facilities are including a large number of these genes in a single panel test using massively parallel (or next generation) sequencing, at considerably reduced cost. However, these gene panel tests pose a considerable challenge to clinical genetic services as many of these genes are not validated as breast cancer susceptibility genes, and even for those that have been, the risks associated with different types of mutations are poorly defined.2 For a test to be useful, the estimated reduction in risk to a mutation carrier should be known, as should the estimated risk of cancer for the non-mutation carriers in the same family.
The genes currently included in commercial breast cancer susceptibility gene panels (in addition to BRCA1 and BRCA2) vary between laboratories and companies, and range from the reasonably well-characterised breast cancer predisposition genes (TP53, PALB2, ATM, CHEK2, CDH1, PTEN, STK11 and one recurring mutation in NBN3), to genes where there is only limited evidence that mutations confer an elevated breast cancer risk (eg, BARD1, BRIP1, MRE11, RAD50, RAD51C, RAD51D, CDKN2A and XRCC2).2 The genes selected for these panels appear to be driven by commercial interests rather than by the strength of evidence for their roles in breast cancer susceptibility. It is vital that we verify which of these are breast cancer susceptibility genes and the magnitude of their associated risks in order to make the best use of data from gene panel testing in clinical genetics service. For carriers of pathogenic mutations in breast cancer susceptibility genes, such as BRCA1 or BRCA2, there are interventions, including mastectomy, bilateral salpingo-oophorectomy and medications, which can be used to reduce risk. However, surgical options are invasive and irreversible, and risk reduction medications may have unfavourable side effects, so clinicians must have robust data with proven utility before using genetic testing involving the new suites of breast cancer susceptibility genes as the basis of risk assessment and risk management. Clinicians also need an understanding of the value of a negative predictive test for other family members once a causative gene mutation in the family has been established.
We sequenced 17 known and putative breast cancer susceptibility genes, plus BRCA1 and BRCA2, in 684 women affected by breast/ovarian cancer and with a strong family history of breast cancer. Protein-truncating and putative deleterious missense mutations were then genotyped in all available family members for assessment of co-segregation of the variant with disease in the family in order to estimate the breast cancer risks associated with these mutations.
Methods
Patient cohort
We selected 684 ‘non-BRCA1/2’ families from the Kathleen Cuningham Foundation Consortium for research into Familial Breast cancer (kConFab). Non-BRCA1/2 families have been recruited into kConFab through Familial Cancer Clinics if they fulfil one of the following criteria1: (i) four or more cases of breast or ovarian cancer on one side of the family, and at least two living affected and four living unaffected family members; (ii) three cases of breast or ovarian cancer on one side of the family including at least one with high-risk features (male breast cancer, bilateral breast cancer, breast plus ovarian cancer in the same woman or breast cancer before age 40), and at least two living affected and four living unaffected family members; or (iii) potentially high-risk individuals from whom fresh tumour is available for research but who do not have at least two living affected and four living unaffected family members.
We selected 684 families from kConFab based on the following criteria: (i) no known pathogenic mutation in BRCA1 or BRCA2 in any family member at the time of selection and (ii) no known protein-truncating mutation in PALB2 or the ATM V2424G mutation. Most, but not all families, had had some BRCA1/2 testing performed prior to selection. In addition, we prioritised family selection (i) on the basis of age of diagnosis of the individual to be sequenced, (ii) including families with a case of pancreatic cancer (n=94), (iii) families who had previously had the most sensitive and complete testing of BRCA1 and BRCA2, (iv) the availability of at least two germline DNA samples from related family members and (v) ranking based on the probability calculated by BOADICEA of carrying a BRCA1/2 mutation. From these families, we selected the youngest breast cancer case for which germline DNA was available as the index case for sequencing.
Targeted sequencing and selection of putative mutations
We performed custom-designed targeted sequencing covering the coding exons of BRCA1, BRCA2, TP53, PALB2, ATM, CHEK2, CDH1, PTEN, STK11, BARD1, BRIP1, MRE11, NBN, RAD50, RAD51C, RAD51D, CDKN2A, CDK4 and XRCC2. Targets were captured using the Agilent Target Enrichment kit by Axeq Technologies according to the manufacturer's instructions and sequenced by 100 bp paired-end reads on the Illumina HiSeq 2000 platform. High-quality sequencing data with average depth of 354.4-fold (ranging from 24.61–1505.14 per exon) was obtained. Sequences were aligned to human reference genome (GRCh37) using BWA.4 Reads marked as PCR or optical duplicates were removed from consideration by Picard (http://picard.sourceforge.net). Variants were called from the targeted exons, and also from the off-target reads up to 25 nucleotides into the introns to capture potential splice-site mutations. Single-nucleotide variants and small indels were called by GATK.5 After calling, to eliminate possible false positive variants, we applied strict quality control filtering excluding variants with <30 reads or variants with reference: alternative reads ratio <0.2 or >5.0.
We used ANNOVAR (http://annovar.openbioinformatics.org) to annotate the functional consequences of variants, as well as to retrieve allele frequencies from the public databases (dbSNP138, 1000 Genomes Project, complete genomes and NHLBI-ESP 6500 exomes).6 The potential functional importance of non-synonymous variants was predicted by the following tools: SIFT,7 PolyPhen2,8 LRT,9 MutationTaster10 and CADD.11 Any non-synonymous variant described by at least four of these algorithms as damaging or probably damaging, or with a CADD score >15, was regarded as putatively deleterious, and selected for segregation analysis. Protein-truncating mutations within the last exon were removed from our candidate list since they are predicted not to activate nonsense-mediated mRNA decay. Impact on splicing was predicted for intronic variants using stand alone Perl scripts of MaxEnt (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html).
Family sequencing and segregation analysis
Segregation analysis for all putative pathogenic mutations was carried out by either Sanger sequencing using the standard sequencing procedure of BigDye Terminator V.3.1 Cycle Sequencing Kit in the PE Applied Biosystem (PE Applied Biosystem) or by Fluidigm 96.96 Dynamic Array integrated fluidic circuits on the BioMark HD System (Fluidigm Corporation) according to the manufacturer's instructions. Primer sequences can be provided upon request.
In order to assess the association of sequenced variants with breast cancer, we performed two analyses based on maximising the likelihood of the observed pedigree genotypes conditional on the pedigree phenotypes and the genotype of the index case. The primary analysis calculated the penetrance for breast cancer in carriers of the susceptibility genes assuming a constant relative risk with age. The second analysis assumed the relative risk to be a constant multiple of the Antoniou et al12 estimates for BRCA2 and calculated the cumulative penetrances at each trial value of the multiplier, allowing for a similar pattern of age-specific effects as in BRCA2, but only required estimation of a single parameter.
Models were fitted under maximum likelihood theory using a modified version of the LINKAGE genetic analysis package.13 Non-carriers were assumed to be at population risks specific to Australia with incidence rates taken from cancer registry data obtained from Cancer Incidence in Five Continents, VIII (IARC, Lyon) and risk ratios (RR, the age-specific breast cancer incidence rate in carriers divided by the relevant population rate) were estimated. Under each model, we first considered all reported cases to be included as affected, and then restricted analyses to cancer cases confirmed by pathology, medical records or death certificates. Reported breast cancers with unknown age at diagnosis were excluded from all analyses. Cancers other than breast (including ovarian cancer) were treated as unaffected at the age of their cancer diagnosis.
We performed two sensitivity analyses, specifically (1) including ovarian cancer as affected and (2) weighting each variant by a predicted deleterious likelihood ratio score based on a weighted function of 10 different in silico analyses as well as the minor allele frequency of the variant. The weights were normalised to sum to the observed number of variants for each gene.
Histopathology
A uropathologist (DC) reviewed one representative H&E-stained section of formalin-fixed, paraffin-embedded tissue from three cases of prostate adenocarcinoma.
Results
In the 684 index cases tested, we identified 11 pathogenic (frameshift or canonical splice site) BRCA1 mutations in 13 individuals and 10 unique BRCA2 mutations in 11 individuals. None of the 24 carriers had previous BRCA1/2 testing. In the remaining 660 non-BRCA1/2 index cases, we found 31 putative deleterious mutations, both protein-truncating and non-synonymous missense, in the other known breast cancer susceptibility genes, in 45 index cases: ATM (n=8; including three missense), CDH1 (n=1; missense), CHEK2 (n=14 families; all truncating and including six unique mutations), PALB2 (n=14 families; eight unique mutations, including two missense), PTEN (n=1; missense), STK11 (n=1; probably splicing) and TP53 (n=6; all missense) (table 1). We also identified 22 potential deleterious mutations in 31 index cases in BARD1 (n=4; including two missense), BRIP1 (n=16; including seven missense), CDK4 (n=1; missense), MRE11 (n=4; missense), NBN (n=1; frameshift), RAD50 (n=1; missense), RAD51C (n=1; missense) and RAD51D (n=3; including two missense), but none in CDKN2A or XRCC2. One intronic variant, c.375–8C>A in STK11, was included because it had an increase in the MaxEnt score of 5.68 compared with the canonical sequence and so was considered a putative splice-site mutation.14 The minimum coverage for any exon sequenced was 25-fold (mean 354-fold), making it very unlikely that we have missed any pathogenic mutations. From the 76 individuals who carried putative pathogenic mutations in genes other than BRCA1 and BRCA2, there were an additional 558 family members with available germline DNA for segregation analysis. All of the 76 mutations were validated in the index case.
Putative mutations found in genes other than BRCA1 and BRCA2
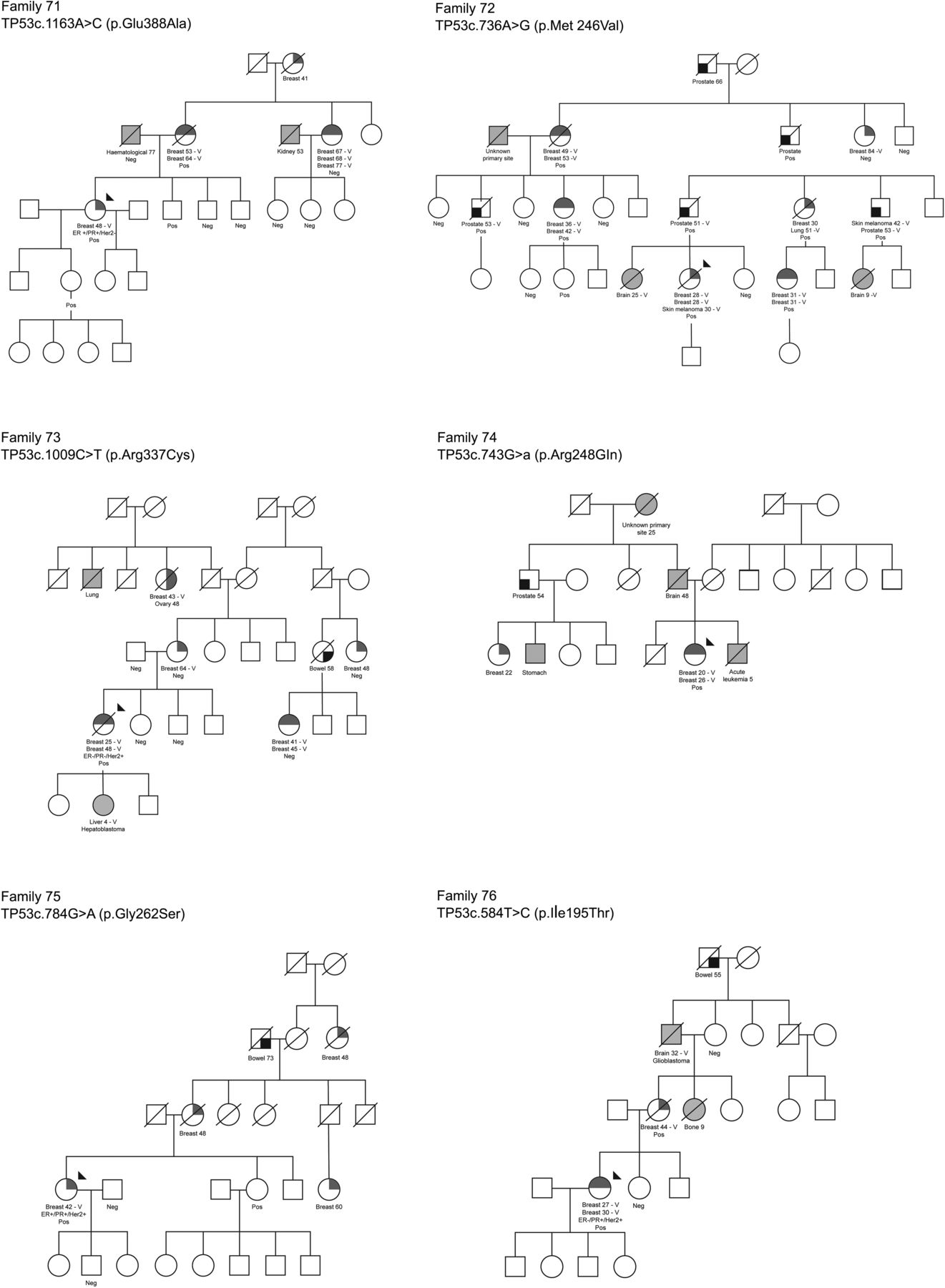
For the primary analysis assuming the constant relative risk with age, only four genes (CDH1, CHEK2, PALB2 and TP53) showed evidence of a significantly increased risk of breast cancer (lower CI >1.0) when the analysis included all cases regardless of validation of their reported diagnoses (table 2). Sections from three cases of adenocarcinoma of prostate occurring in carriers of p.Met246Val TP53 in family 72 were reviewed. All three cases showed high-grade carcinoma with Gleason score 9. Two cases had associated intraductal carcinoma of prostate (IDC-P) while the third case had a component of large cribriform carcinoma, although no IDC-P was demonstrated in the section examined.
Risk ratios for each of the genes examined
There was some evidence of an association between ATM and breast cancer risk (RR=2.51; 95% CI 0.95 to 8.26 when including all cancer cases; or RR=2.67; 95% CI 0.82 to 10.56 when restricting analysis to cases with a diagnosis confirmed through medical records or cancer registry matching), but this association was not significant. For BRIP1 (RR=0.47; 95% CI 0.15 to 1.18), any clinically meaningful increased risk of twofold could be excluded, given the upper CI limit of 1.18 for the most informative analyses using all cases; if we consider only cases with a confirmed diagnosis, HRs >1.56 can be excluded.
Some of the genes in the set have been associated with ovarian cancer risk.15–18 However, including ovarian cancers in our analyses did not substantially change the results, largely due to the small number of verified and unverified ovarian cases in these pedigrees, which also prohibited estimation of ovarian-specific risks. In particular, including ovarian cancer as affected did not change the estimates for either BRIP1 or RAD51C (data not shown). Weighting by the probability of pathogenicity of each variant using the composite scores from the PERCH suite of programs (http://www.fengbjlab.org/perch) also did not have a large effect on the results. For ATM, for example, the weighted RR was 2.55 (95% CI 0.95 to 8.30) compared with unweighted RR of 2.51 (95% CI 0.95 to 8.26).
Discussion
Several recent publications have described panel testing of known and putative breast cancer genes in early-onset or multiple-case breast cancer families,19–23 or unselected series of triple-negative breast cancer cases.24 However, none of these studies had access to multiple DNA samples from the carrier families for segregation analysis. They were, therefore, unable to estimate the risks associated with mutations in any of the putative breast cancer susceptibility genes, but limited their conclusions to the frequency of these mutations, and the impact of these findings on clinical care. We tested 17 putative breast cancer susceptibility genes in 684 index cases with a strong family history of breast cancer (660 of which did not carry BRCA1 or BRCA2 mutations), which makes it one of the largest studies of these genes in breast cancer families to date. Furthermore, we were able to perform segregation analysis of the 76 putative deleterious mutations found because of the large numbers of blood samples (mean 7.3 per family) collected by kConFab. We found that mutations in these Australasian families in CHEK2, PALB2 and TP53 conferred significant risks of breast cancer and also found some support for a moderate risk of breast cancer associated with mutations in ATM. In addition, we found a single family with a p.Asp777Asn CDH1 mutation (RR=13.62; 95% CI 1.12 to 24.0), which suggests that CDH1 is a breast cancer susceptibility gene (figure 1). We found no such supporting evidence for BARD1, BRIP1, CDKN2A, CDK4, MRE11, NBN, RAD50, RAD51C, RAD51D, STK11 or XRCC2 as moderate-risk or high-risk breast cancer susceptibility genes. However, it should be noted that for all the genes we evaluated except for BRIP1 the upper limit of our 95% CIs does not exclude the possibility that these genes confer at least a moderate risk of breast cancer. Two of the 13 families identified with missense mutations in BRIP1 also had pathogenic mutations in BRCA2.

{kind=link}
{kind=link}
Pedigrees of the families with mutations in TP53, CDH1, NBM and RAD51C, and a representative pedigree of a family with PALB2 p.Trp1038*. Neg, non-carrier; Pos, mutation carrier.
A recent review of the literature2 concluded that protein-truncating mutations in BRCA1 and BRCA2, probably in PALB2 and perhaps also in PTEN, confer high risk (defined as more than fourfold) of breast cancer but that only a small minority of missense variants in BRCA1 and BRCA2 are associated with high risks for breast cancer. Although our point estimate for risk associated with PALB2 variants is less than threefold, the 95% confidence limits (ninefold) on that estimate do not exclude the possibility that this is a high-risk breast cancer susceptibility gene. Although our risk estimate is not consistent with the very high estimate previously reported in Australian families,25 the 95% confidence limits from both analyses overlap; similarly, our 95% confidence limits overlap those of the published meta-analysis (3–9.4).26 In addition, both missense and protein-truncating variants in TP53 are associated with substantially increased risks for breast cancer. Protein-truncating mutations in CHEK2, as well as protein-truncating and some missense mutations in ATM, are considered to be moderate risk (2–4-fold) breast cancer susceptibility genes. There are three genes (STK11,27 CDH128 ,29 and NBN3) with clear evidence for association for at least some mutations, but for which the risk estimates are too imprecise to describe them as high-risk or moderate-risk breast cancer susceptibility genes. For CDH1 and STK11, the finding of a mutation in a family with hereditary diffuse gastric cancer or Peutz–Jegher syndrome (PJS) respectively, provides a syndrome-associated mutation where the risk of breast cancer is well established and predictive genetic testing usually proceeds within that family. However, when such a mutation is identified outside that clinical context, such as a STK11 mutation in a family with breast cancer only and no clinical sign of PJS, the breast cancer risks associated with that mutation are unclear and the value of predictive testing remains uncertain. However, we should note that the STK11 mutation we report is only predicted to be pathogenic by in silico analysis; functional conformation would be required to confirm this but no cell line is available for splicing assays. For NBN, the data are extremely limited and most data apply only to one specific mutation within one population.30 For all the other genes that have been described as breast cancer susceptibility genes (BARD1, BRIP1, CDKN2A, MEN1, MLH1, MRE11, MSH2, MSH6, NB1, PMS2, RAD50, RAD51C, RAD51D and XRCC2), the review found insufficient evidence to describe them as such.2 In combination with our own data, we conclude that there is no clinical value in mutation testing these 11 genes in breast cancer families because there is no evidence that mutations in them confer twofold or greater risks of breast cancer.
The risks we have estimated for ATM, CARD1, BRIP1, MRE11, PALB2, RAD51D and TP53 could be underestimated due to a mixture of pathogenic and non-pathogenic variants. Functional assays of the missense variants are needed to identify those most likely to be pathogenic. However, when weighting the results by the probability of pathogenicity, the results did not change significantly compared with the unadjusted, probably due to the relatively small range of posterior probabilities of pathogenicity caused by the selection of variants predicted to be deleterious by a subset of the in silico algorithms used in the computation of the weights. The number of variants in each gene was not large enough to analyse the data using a heterogeneity model, which assumes that the variants are a mixture of those that confer no increased risk and true pathogenic variants.
Figure 1 shows the six pedigrees with TP53 mutations and one with a CDH1 mutation (in which only one of the breast cancer cases had lobular morphology), and an illustrative PALB2-mutated pedigree, as well those with RAD51C and NBN mutations, which did not segregate with breast cancer-affected status. Review of the only available case of breast cancer in a CDH1 mutation carrier showed the carcinoma had mixed ductal and lobular features, with reduced and aberrant staining for E-cadherin in the lobular regions. Only two of the six TP53 families (families 74 and 76) fulfilled the criteria for Li Fraumeni Syndrome (LFS), and one (family 73) appeared to derive from a de novo mutation. Five of the six missense mutations (excluding p.Glu388Ala) found in TP53 have been previously identified as somatic or germline mutations in the IARC database,31 but both p.Glu388Ala (family 71) and p.Gly262Ser (family 75) are regarded as unclassified variants by Clin Var (http://www.ncbi.nlm.nih.gov/clinvar/). Interestingly all of the four breast cancer cases for which pathology data were available were ER+, PR+ and/or HER2+, consistent with the report from Masciari et al32 that most breast tumours in LFS are hormone receptor and/or HER2 positive. One family (family 72) contained four cases of adenocarcinoma of the prostate who were all carriers or obligate carriers of the TP53 mutation (p.Met246Val), diagnosed at ages 51, 53, 53 and 66. Although prostate cancer is not a recognised feature of LFS, this family is so remarkable that we performed pathology review. Sections were reviewed from three cases of prostatic adenocarcinoma occurring in men with the p.Met246Val TP53 mutation, and both cases showed high grade, Gleason score 9 carcinoma, and one case had IDC-P. Interestingly, these three tumours resemble prostatic adeonocarcinomas from men with germline mutations of BRCA2, which are associated with poor survival rates,33 and suggest that further analysis of TP53 mutation status, as well as histopathology and survival analyses, would be warranted in prostatic adenocarcinoma.
We identified three families with rare, evolutionarily unlikely missense mutations in the FAT or kinase domains of ATM (p.Ala2274Thr, p.Asp2720His and p.Val2424Gly), which are predicted to confer increased risks of breast cancer.34 The single family we identified with a mutation in PTEN (p.Arg142Trp) did not have any clinical signs of Cowden syndrome, but the majority of mutations in Cowden syndrome are protein truncating35 ,36 so p.Arg142Trp should be described as an unclassified variant. The missense variants we found in the known breast cancer susceptibility gene, PALB2, should also be considered unclassified variants because there is no evidence at present that they are associated with elevated breast cancer risk.
In conclusion, by panel testing 17 known or putative breast cancer susceptibility genes in 660 non-BRCA1/2 multiple case breast cancer families we found 10 mutations that were clinically notifiable in Australia—one missense (p.Val2424Gly) in ATM, five families with the p.Trp1038* truncating mutation in PALB2 and four missense mutations in TP53—demonstrating the clinical value from such findings. At present, all Australian clinics agree to notify families when a PALB2 c.3113G>A (p.Trp1038*) mutation is found as the breast cancer risk associated with this particular mutation is more firmly established and known to approach that attributed to BRCA2. Once the risks attributable to truncating PALB2 mutations outside that specific context are clear, further families may be notified about other mutations, but the general approach in Australia has been conservative thus far. While most of the families with a TP53 mutation have features typical of the LFS, others would not have been detected through family history alone. In these families, the finding of a pathogenic TP53 mutation allows for a change of management in that young unaffected female mutation carriers may elect for risk-reducing bilateral mastectomy (or bilateral mastectomy as opposed to breast conservation treatment if diagnosed with breast cancer in the future). TP53 carriers may also wish to participate in other cancer screening programmes available in research studies. It also allows for the possibility of family planning using prenatal testing or preimplantation genetic diagnosis. Finally, the family members who do not carry the causative TP53 mutation are relieved of a very high risk of multiorgan cancers for themselves and their offspring. In families with the PALB2 c.3113G>A mutation, carriers have a risk of breast cancer that approximates that related to a BRCA2 gene mutation, so mutation carriers can take advantage of additional breast cancer screening (including MRI), risk-reducing medications or surgery. Similar management applies for women in families with the high-risk ATM variant (p.Val2424Gly). Although a high ovarian cancer risk has been associated with BRCA1/2 mutations, this is not so for either PALB2 or ATM, so women from these families can be reassured about ovarian cancer risk. Although the c.1100delC mutation in CHEK2 confers a moderate risk of breast cancer, the clinical utility of testing other family members for this variant is unknown.
Overall, additional panel testing for the non-BRCA1/2 families provided additional important and new clinical information for <2% of breast/ovarian cancer families, so while panel testing is becoming increasingly popular, there is limited value to be gained from such a very broad approach until further data are available to guide the management of the large majority of families whose genetic susceptibility will not be resolved by panel testing. There is also the potential for families to overinterpret the clinical relevance of variants in the less-well-validated putative predisposition genes included in these panels, with the consequence that they may be tempted into radical measures to reduce breast cancer risk or alternatively for family members to abandon effective cancer risk management options if they do not carry variant carried by the index case. Although we have shown that segregation analysis has some potential to determine breast cancer risks associated with mutations in known and putative breast cancer susceptibility genes, very large case–control studies and/or large family-based studies are needed to define the risks more accurately.
Acknowledgments
We thank Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study for their contributions to this resource, and the many families who contribute to kConFab.
References
Footnotes
Contributors DG, HT and GC-T selected the families for analysis. IM designed the exon capture. JL and JE did the sequence alignment and selected the putative pathogenic mutations. SH and HT performed the sequencing of family members. HM, B-JF and DG performed the segregation analysis. IC and MS helped to fund the study. DC reviewed the prostate adenocarcinoma cases. GM and JK provided clinical input. JL and GC-T wrote the manuscript, with input from all authors.
Funding This project has been supported by funding from the Susan Komen Foundation as well as the National Health and Medical Research Council. The Clinical Follow Up Study has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia and the National Institute of Health (USA). kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. HM is supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research and the Ministère de l'enseignement supé- rieur, de la recherche, de la science et de la technologie du Québec through Génome Québec as part of the PERSPECTIVE project (CIHR grant GPH-129344) . This work was supported in part by NIH grants CA128978 and CA116167.
Competing interests DG has US Patent nos. 5747282; 5710001; 5837942; 6033857 with royalties paid by Myriad Genetics. GM reports personal fees from AstraZeneca, outside the submitted work.
Patient consent Obtained.
Ethics approval IRB Peter MacCallum Cancer Centre (Victoria, Australia).
Provenance and peer review Not commissioned; externally peer reviewed.