Article Text

Abstract
Background Phenotypic overlap among the inherited bone marrow failure syndromes (IBMFSs) frequently limits the ability to establish a diagnosis based solely on clinical features. >70 IBMFS genes have been identified, which often renders genetic testing prolonged and costly. Since correct diagnosis, treatment and cancer surveillance often depend on identifying the mutated gene, strategies that enable timely genotyping are essential.
Methods To overcome these challenges, we developed a next-generation sequencing assay to analyse a panel of 72 known IBMFS genes. Cases fulfilling the clinical diagnostic criteria of an IBMFS but without identified causal genotypes were included.
Results The assay was validated by detecting 52 variants previously found by Sanger sequencing. A total of 158 patients with unknown mutations were studied. Of 75 patients with known IBMFS categories (eg, Fanconi anaemia), 59% had causal mutations. Among 83 patients with unclassified IBMFSs, we found causal mutations and established the diagnosis in 18% of the patients. The assay detected mutant genes that had not previously been reported to be associated with the patient phenotypes. In other cases, the assay led to amendments of diagnoses. In 20% of genotype cases, the results indicated a cancer surveillance programme.
Conclusions The novel assay is efficient, accurate and has a major impact on patient care.
- Clinical genetics
- Diagnostics tests
- Haematology (incl Blood transfusion)
- Other oncology
Statistics from Altmetric.com
Introduction
Inherited bone marrow failure syndromes (IBMFSs) are multisystem disorders with underproductive bone marrow and single-lineage or multilineage cytopenia.1 Many of the disorders carry a risk of cancer. The term IBMFSs is reserved for disorders that are caused by germline mutations, either inherited or arising de novo with the patient.2 ,3 Based on transmission patterns (eg, autosomal dominant (AD), autosomal recessive (AR) or X-linked), segregation of alleles within families and molecular analysis of IBMFS genes, all known IBMFSs appear to be monogenic.4–6
The wide range of physical anomalies associated with the IBMFSs help establish a specific diagnosis. However, the substantial phenotypic overlap among the disorders frequently limits the ability to establish a diagnosis based solely on clinical manifestations. Furthermore, physical malformations may be absent or appear later in life.7 Identifying the specific mutated gene is essential. It helps establish a diagnosis, predict disease course (eg, cancer risk), direct genetic counselling and treatment and select healthy sibling donors for haematopoietic stem cell transplantation (HSCT). Since >70 IBMFS genes have been identified, timely and cost-effective strategies for genetic testing are necessary to provide proper care.
Sanger sequencing often poses significant limitations during the diagnostic evaluation of patients who have or suspected to have IBMFSs. First, there are >25 defined IBMFSs with substantial clinical overlap among as well as between IBMFSs and acquired marrow failure syndromes. Second, individual diseases (eg, Fanconi anaemia (FA), dyskeratosis congenita (DC) and Diamond–Blackfan anaemia (DBA)) can be caused by mutations in multiple genes. Since Sanger sequencing of multiple IBMFS genes is costly and lengthy,8 it is not feasible in the setting of acute illnesses (eg, presentation with severe aplastic anaemia (SAA)) before urgent treatment decisions are made.
Next-generation sequencing (NGS) generates data on multiple DNA fragments in a single reaction.9 Although application of NGS gene panels has been reported in several heterogeneous disease groups,10–14 there are insufficient data about the clinical impact of this genetic approach on disorders with cancer risk with regard to facilitating a diagnosis, delivery of care and counselling of patients and other family members. There is one published study that used an NGS gene panel to study a variety of IBMFSs.15 In that study that included a mixed population of paediatric and adult patients with bone marrow failure (including IBMFSs) and myelodysplastic syndromes (MDSs), the mutation detection rate was only 11%. We hypothesise that an NGS gene panel, which includes all known genes for single and multilineage cytopenias and uses the HaloPlex technology, could form the basis of a comprehensive testing strategy for IBMFSs in order to provide accurate and clinically relevant molecular diagnoses in a timely fashion and at a significantly reduced cost.
We developed an NGS assay to sequence a panel of 72 known IBMFS genes related to disorders with pancytopenia (eg, FA), disorders with predominantly anaemia (eg, DBA), disorders with predominantly neutropenia (eg, Kostmann/severe congenital neutropenia (K/SCN)), disorders with predominately thrombocytopenia (eg, familial thrombocytopenia (FT)) and inherited MDS (eg, Emberger syndrome). We applied the assay to a large population of 158 patients with IBMFSs. We first assessed the ability of the assay to identify causal mutations in patients that had been diagnosed with a specific IBMFS (eg, FA, DBA, K/SCN and FT), but whose genotype had not been identified. We then tested the ability of the assay to identify genotypes and establish a diagnosis in patients with unclassified IBMFSs.
Methods
Patients
The Canadian Inherited Marrow Failure Registry (CIMFR) is a multicentre study, which was approved by the Institutional Ethics Board of all 17 participating institutions. In Canada, all paediatric patients with IBMFSs are typically treated in one of the CIMFR institutions. This registry is population-based as >90% of the patients in this study are from centres that enrol >80% of the patients at their institutions. Patients have been prospectively enrolled since January 2001 after obtaining written consent. Detailed information was collected at presentation, study entry and periodic follow-up.
Individuals who fulfilled published diagnostic criteria for an IBMFS5 were recruited at the participating centres of the CIMFR. In short, the eligibility criteria include evidence of chronic bone marrow failure in addition to either family history or physical malformations or presentation earlier than 1 year of age or positive genetic testing. When possible, each case was assigned a specific syndromic diagnosis by the participating centre. Diagnoses were reviewed centrally and if necessary were adjusted based on published diagnostic criteria of specific IBMFSs2 ,5 and after discussions with the respective centre. Cases that fulfilled the diagnostic criteria of an IBMFS but did not meet the clinical, laboratory and genetic diagnostic criteria for any known IBMFS2 ,5 were defined unclassified IBMFSs.8 The majority of these patients had undergone extensive genetic testing, which was negative. Patients who presented with SAA and did not respond to immunosuppressive therapy (ie, multilineage severe cytopenia and marrow cellularity <25% at 3 months after starting therapy) were also eligible for the CIMFR as having unclassified IBMFSs since a proportion of such patients would be ultimately diagnosed with IBMFS.
In the present study, 158 patients with classified or unclassified IBMFS without identified causal mutations were included; 155 were enrolled in the CIMFR and 3 were enrolled in an internal bone marrow failure study at the Hospital for Sick Children, Toronto. Of the 158 patients, 69 had prior clinical genetic testing, 72 were not tested and 17 had no available data on previous testing.
NGS IBMFS gene panel assay
We designed an NGS assay for a comprehensive panel of 72 IBMFS genes that had been published as of January 2013 (see online supplementary table S1). DNA was extracted from peripheral blood in most cases. For patients with MDS/acute myeloid leukaemia (AML), skin fibroblasts, marrow fibroblasts or peripheral blood T-cells were used for DNA extraction to minimise detection of somatic changes. We used the HaloPlex Capture Kit (Agilent Technologies, Santa Clara, California, USA) for DNA library preparation and capture according to the manufacturer's instructions. Targeted fragments were amplified while incorporating indexes and generating linear barcoded DNA fragments, and were sequenced on the Illumina HiSeq2500 platform. DNA libraries from 83 patients (first batch) and 85 patients (second batch) were pooled, labelled with different barcodes and sequenced in one lane.
Variant analysis and filtering strategy
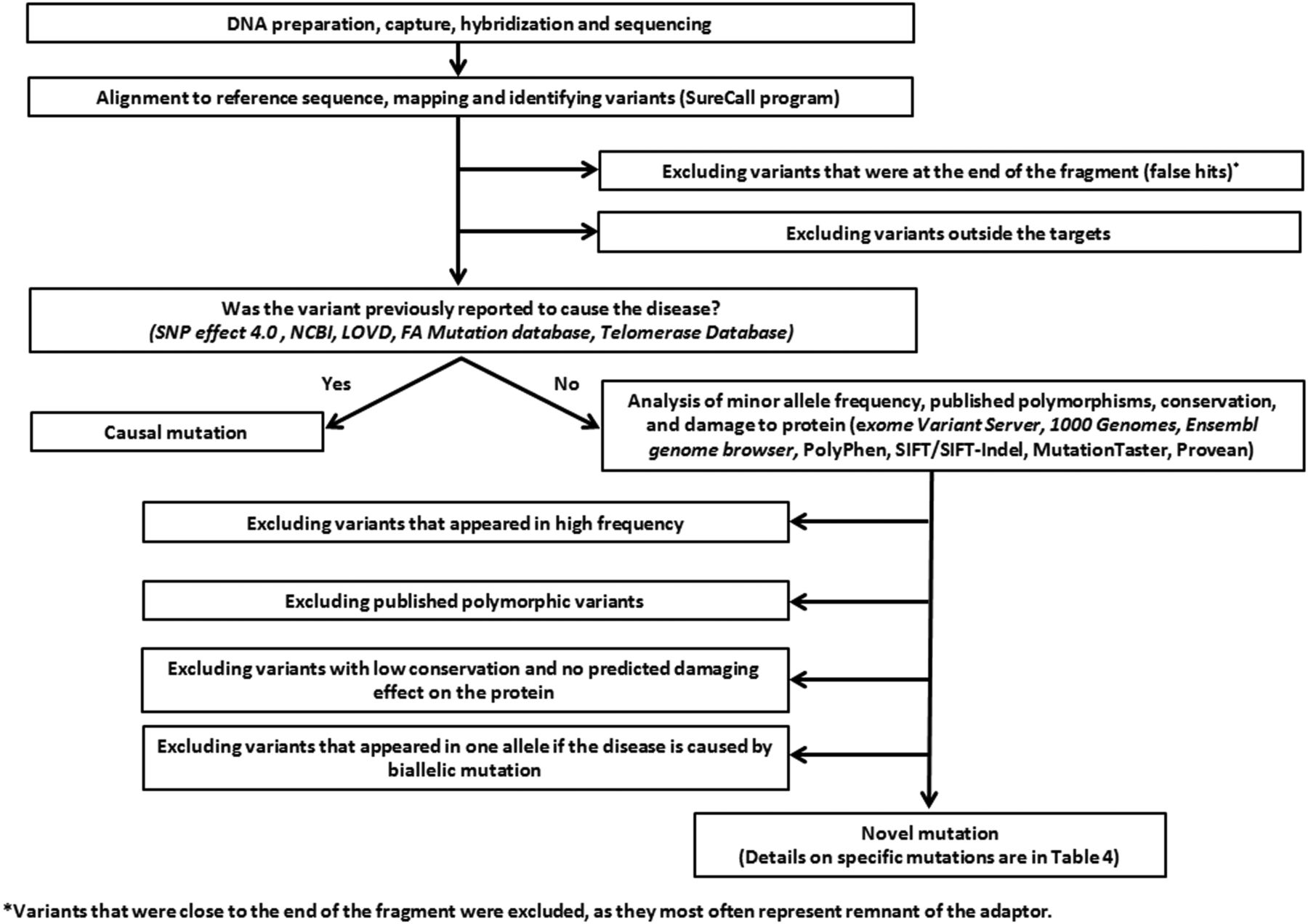
The algorithm used to filter non-relevant variants is described in figure 1. To minimise false positive calls, we considered variants as true hits if they had ≥5 positive reads. For heterozygous calls, we also required that positive reads constitute ≥20% of the total reads for the respective nucleotides. However, to study the ability of the assay to detect mutations, we performed Sanger sequencing of any homozygous variants that appeared in ≥2 reads and any heterozygous variants that appeared in ≥20% of the reads. The software programs used to study minor allele frequency (MAF), conservation and potential damage of variants on the protein are listed in table 1.
Software that were used in this study to evaluate conservation, minor allele frequency and potential damage of variants on splicing and protein structure and function

An algorithm summarising analysis and filtering processes. FA, Fanconi anaemia; LOVD, Leiden Open Variation Database; NCBI, National Center for Biotechnology Information; PolyPhen, polymorphism phenotyping; SIFT, sorting intolerant from tolerant.
Variants were defined as ‘causal’ if they had been reported as disease-causing in public databases (table 1). Novel variants were considered ‘most likely causal mutations’ if (1) they appeared in allelic dosage that was consistent with the known inheritance mode of the disease, (2) the MAF was <0.001, (3) evolutionary conserved amino acid/s are affected and (4) the variant was considered damaging by at least two of the following prediction software programs: PolyPhen2, SIFT/SIFT-Indel, Provean, MutationTaster and Human Splicing Finder. In this paper, we referred to both previously published mutations and novel mutations as ‘causal mutations’.
Previously reported variants were considered ‘not causal’ if they had been published to be polymorphic. Novel variants that did not fulfil the criteria in the previous paragraph were deemed ‘most likely not causal’. Variants that fulfilled most but not all the above criteria remained of unknown significance.
Sanger sequencing
Targeted sequences were analysed after PCR amplification by bi-directional sequencing as previously described.16
Statistical analysis and calculation of test sensitivity and precision
Calculation of test sensitivity and precision was performed as previously described.17
Sanger sequencing was considered as the reference standard method. Assay sensitivity was determined as ∑ true positive/∑ condition position, where ‘true positive’ was considered when a mutation was observed by both NGS and Sanger sequencing and ‘condition positive’ was considered in any case where a mutation was observed by Sanger sequencing. Assay precision was determined as ∑ true positive/∑ test outcome positive, where ‘test outcome positive’ was considered as any case with an identified causal mutation by NGS.
Results
Frequencies of variants and causal mutations
The assay covered 456 351 bp. The average gene coverage was 99.12% (see online supplementary table S1). The average read depth was 680×. Also, 91.2% of targeted regions were covered with >100×. Among the 158 patients without known causal mutations, we identified 66 393 variants. After filtering (figure 1), 77 nucleotide-level variants (mutant alleles) in 59 patients were deemed causal (tables 2 and 3; see online supplementary tables S3 and S4). The majority of the mutations (44) have previously been reported; 33 were novel (see online supplementary table S5). Of the novel mutations, 15 were splicing mutations or indels, and 18 were missense mutations. Three of the novel mutations recurred in more than one patient.
Genotyping patients with classified IBMFSs and unknown genes
Genotyping of patients with unclassified inherited bone marrow failure syndromes
Efficiency of variant detection
We evaluated the ability of the assay to detect 53 variants that were found by clinical testing prior to the present study; 40 were polymorphisms in 21 of the 158 subjects in this study, and 13 were causal mutations in 10 other patients on the registry that had been genotyped (see online supplementary table S2). All variants were detected by the NGS assay, except for one polymorphic variant that was not covered; yielding a sensitivity of 98%.
Next, we determined the ability of the assay to detect mutations with sizable number of reads, which we defined as homozygous mutations of ≥5 reads and heterozygous mutations that appear in ≥5 reads and constitute ≥20% of the total reads. All 76 identified such causal mutations were validated by Sanger sequencing, giving a precision of 100% (see online supplementary figure S1–S58). Next, we studied the ability of the NGS assay to detect mutation calls with <5 reads. Among three such calls, two were found true by Sanger sequencing (see online supplementary figures S10 and S22). This suggests that calls with few reads may still be true and require validation.
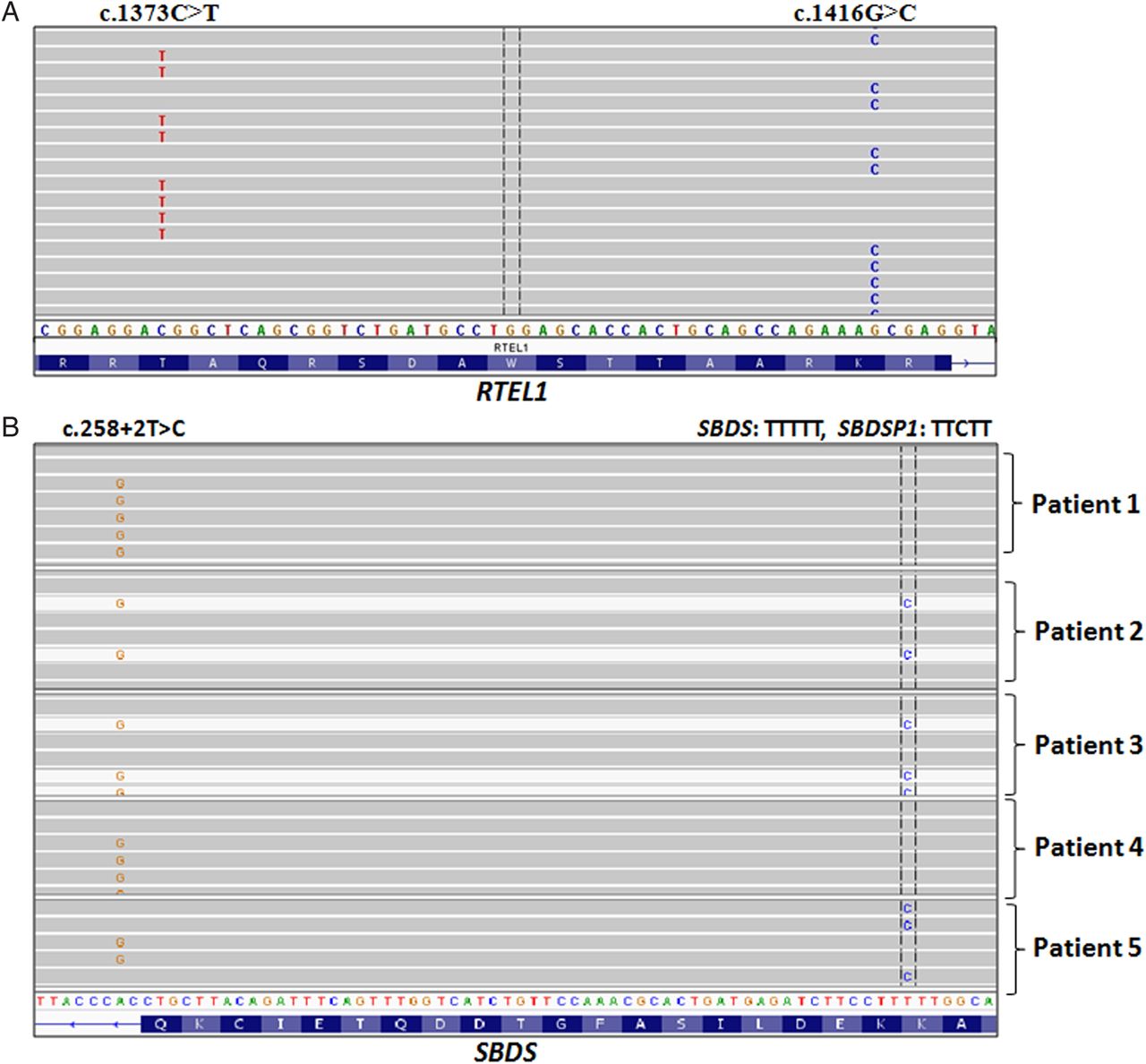
We encountered several cases where the NGS assay outperformed Sanger sequencing in assessing complex genotypes. For example, the assay enabled determination whether two mutations in RTEL1 were on the same allele (figure 2A), and whether a mutation in SBDS is true and not a contaminating pseudogene sequence (figure 2B).

Next-generation sequencing (NGS) inherited bone marrow failure syndromes gene panel assay revealed challenging genotypes. (A) Using BAM files to depict results of massive parallel sequencing, each of the obtained sequences of RTEL1 (see online supplementary table S4, patient 13) could be visualised separately. Both variants are located closer than the 150 bp size of the oligonucleotides used and it is possible to determine that they are on different alleles (and read). (B) SBDS and its pseudogene (SBDSP1) share 97% homology. Both were amplified by the designed NGS oligonucleotide. Sequences from several patients with the c.258+2T>C variant are seen. This common SDS variant results from conversion with SBDSP1. Based on the difference between SBDS sequences upstream to the mutation (TTTTT) and SBDSP1 (TTCTT), we can discriminate true mutation from a contaminating SBDSP1 sequence. Patients 1 and 4 have c.258+2T>C variant together with the SBDS sequence TTTTT, therefore, those variants are real SBDS mutations. Patients 2 and 3 have c.258+2T>C variant together with the SBDSP1 sequence, TTCTT, indicating that these sequences are of SBDSP1 sequences and not real SBDS mutations. Patient 5 has both, c.258+2T>C variant and TTCTT, but they exist in different fragments, indicating that both are real SBDS mutations, and that they exist in different alleles.
Genotyping patients with classified IBMFSs
Of the 75 IBMFS patients with clinically classified IBMFSs (table 2), we identified 60 nucleotide-level causal mutations (9 of them were homozygous) in 44 patients (59%) by the NGS assay (see online supplementary table S3). Among patients who had not had previous genetic testing, 66% were genotyped. DBA was the most common IBMFS in the Canadian registry, followed by FA; among these disorders, 70% and 75% were found to have causal mutations by the NGS assay, respectively.
The NGS assay helped establish a precise diagnosis and discriminate between disorders with similar initial phenotypes but different natural histories. For example, one of two patients with FT (see online supplementary table S3, patients 38 and 39) had a mutation in MYH9, leading to a specific diagnosis of MYH9-associated FT, while the other was classified as having ANKRD26-associated FT, based on having a frameshift mutation in the ANKRD26 gene. This frameshift causes loss of the last 50 amino acids of the protein, a region that is critical for the binding of ANKRD26 to its partner, TRIO, that shares cellular functions with ANKRD26.18 Importantly, in contrast to MYH9-associated FT, ANKRD26-associated FT is associated with an increased risk of haematological malignancies19 and indicates cancer surveillance.20
The analysis of two cases of thrombocytopenia with absent radii (TAR) syndrome exemplifies how compound heterozygosity that includes both one allelic deletion and a nucleotide-level mutation can be detected by NGS. Such a compound heterozygosity in RBM8A is the commonest cause of TAR syndrome.6 ,21 The NGS data indicated a previously reported mutation in 5′-UTR area of RBM8A in both patients (figure 3A, see online supplementary table S3, patients 43 and 44). To determine whether the patients have homozygous mutations or compound heterozygosity with a submicroscopic monoallelic deletion, we used the SureCall CNV detection algorithm and our NGS data. Comparison of read numbers along RBM8A in these patients with five other subjects (figure 3B) suggested one allelic deletion in both patients. Copy number analysis in one of these cases by Affymetrix SNP6.0 array validated a submicroscopic deletion (figure 3C).

Revealing deletions in the RBM8A gene. (A) A previously described missense mutation in the RBM8A gene: c.-21G>A was detected by the NGS data (upper panel) and validated by Sanger sequencing (lower panel). Only a mutant allele was detected, which suggests either a homozygous mutation or a nucleotide-level mutation that is accompanied by a deletion on the other allele. (B) Copy number analysis was detected in the RBM8A gene. Patient read numbers in the gene region obtained from the NGS data were compared with read numbers from five subjects by the SureCall software. One copy number deletion was found as indicated by log2 of <0.6 when patient calls are compared with each of the other controls. (C) One copy number deletion in the RBM8A gene was validated by the Affymetrix SNP6.0 array.
Amendment of diagnoses
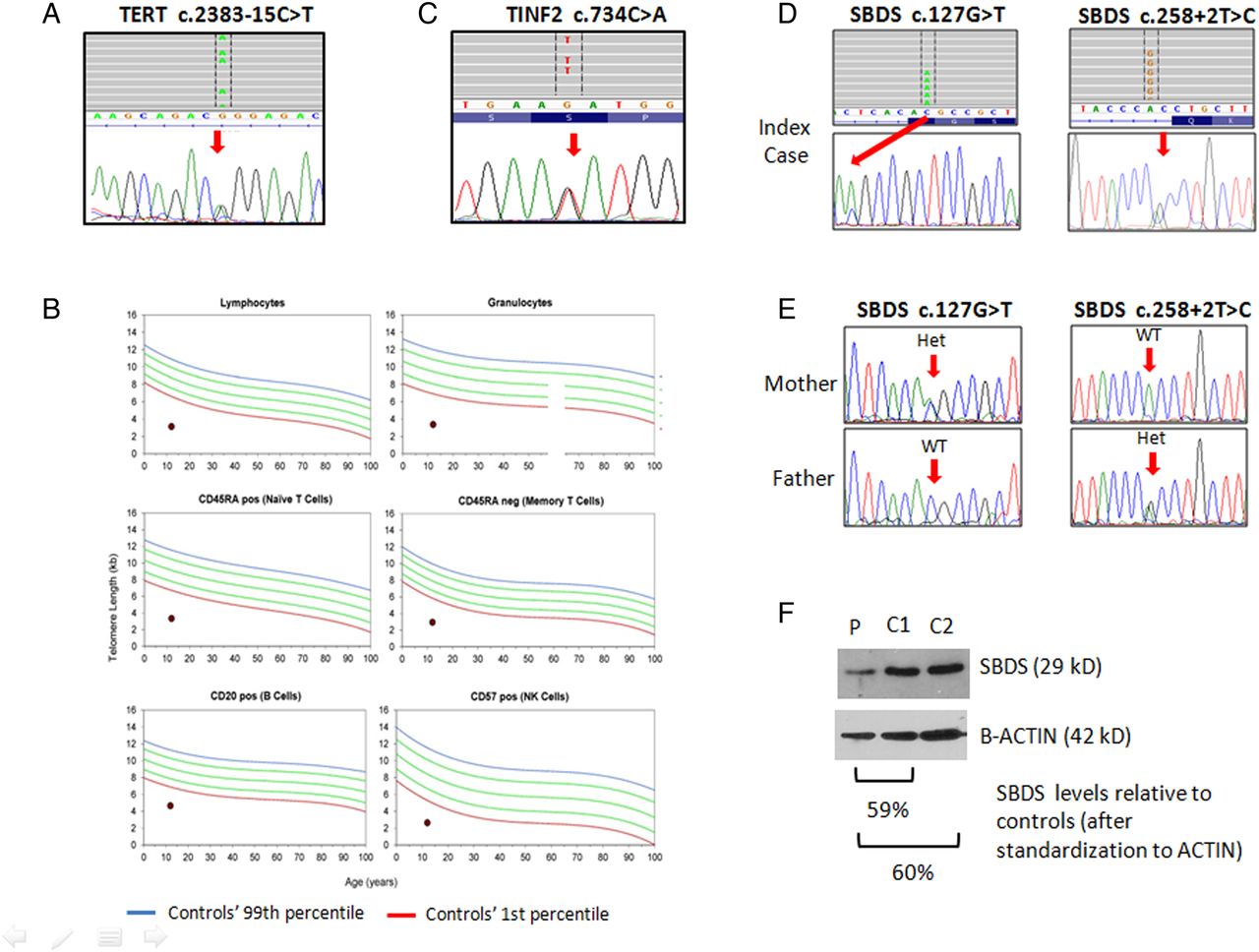
The diagnosis of four clinically classified patients (9%) was amended after the results of the NGS gene panel assay became available. The first example is of a mother and son who were clinically diagnosed with non-syndromic FT (see online supplementary table S3, patients 40 and 41). Both were found to be heterozygous for a TERT mutation c.2383-15T>C (figure 4A), which was predicted to disrupt the binding site of splicing factor SRp40 and break the adjacent splicing site (see online supplementary table S5, patient 9). This mutation appeared in another unrelated patient in our registry, who had aplastic anaemia, very short telomeres in the range that is characteristic to DC and response to androgen therapy (figure 4B, see online supplementary table S2, patient 9). This mutation is very rare in the general population. Based on this information, the diagnosis was changed to DC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mutations that lead to amendment of diagnosis. (A) Heterozygous mutation in TERT, c.2383-15C>T, was identified in a mother and son who were clinically diagnosed as autosomal-dominant non-syndromic familial thrombocytopenia (see also online supplementary table S3, patient 40). Bone marrow biopsy at the age of 17 years showed cellularity of 70%. The upper panel shows the nucleotide-level sequence by the BAM next-generation sequencing (NGS) file, which is the DNA reverse-complement counterpart. The lower panel shows the results of Sanger sequencing. (B) Telomere length in various peripheral blood cell lineages from a with TERT mutations c.2383-15C>T in the study was measured by flow fluorescent in situ hybridisation. The dots represent the patient values. The lines represent percentiles of telomere length among 391 healthy control persons ranging from birth to 100 years of age done in Repeat Diagnostic Laboratory (Vancouver, Canada). (C) TINF2 mutation in a patient who was clinically diagnosed with Fanconi anaemia (FA). The upper panel shows the nucleotide-level sequence as found by NGS. The lower panel shows the results of Sanger sequencing. The patient underwent haplo-identical haematopoietic stem cell transplantation from her parent with FA-adjusted conditioning, but did not engraft. The patient had also a novel germline RUNX1 variant in the marrow fibroblast DNA that was tested (c.383T>C, V128A), which might have contributed to her disease course. (D) SBDS mutations in a patient that was diagnosed clinically with Diamond–Blackfan anaemia. The upper panel shows the nucleotide sequence as found by next-generation sequencing. The middle panel shows the results of Sanger sequencing. (E) Sanger sequencing of SBDS in parents’ samples. (F) for western blotting of the SBDS protein in the patient with SBDS mutations: c.127G>T/SBDS c.258+2T>C (P) and two controls (C1, C2) (C, control; P, patient).
The third patient was diagnosed clinically with FA based on haematological findings noticed at the age of 12.5 months, non-haematological features and increased chromosome fragility with hypersensitivity to mitomycin C and diepoxybutane (see online supplementary table S3, patient 25). Using the NGS assay, we found a mutation in TINF2 (c.734C>A) (figure 4C), which was previously reported in a patient with aplastic anaemia. Accordingly, the diagnosis was amended to DC. Varying degree of chromosomal instability has been reported in DC,22–25 but not to the degree found in this patient. Telomere length measurement was not available before the patient died.
The fourth patient was diagnosed with DBA based on severe anaemia, reticulocytopenia and markedly reduced marrow erythrocytes (see online supplementary table S3, patient 16). Marrow cellularity was reduced for the patient's age (70%), and moderate neutropenia (0.69×109/L) was registered once. The patient failed to respond to steroids. We found two mutations in SBDS (figure 4D): c.258+2T>C, which is the most common SBDS mutation, and c.127G>T, which is predicted to replace valine with leucine. Importantly, the substitution of G at the second last nucleotide of exon 1 (c.127) is also predicted to break the adjacent splice donor site. Sequencing of samples from the patient's parents showed that each parent was heterozygous for one of these mutations (figure 4E), confirming the biallelic nature of the maturations in the child. Sequencing of reverse transcription PCR products using three different pairs of primers repeatedly showed multiple cDNA products consisting with alternative splicing by both mutations. The overall SBDS protein was reduced to 60% of the level in normal samples (figure 4F). Based on the above, a diagnosis of SDS was deemed more likely than DBA.
Identifying mutations and establishing diagnoses in patients with unclassified IBMFSs
Eighty-three patients with unclassified IBMFSs were studied. These patients posed major diagnostic dilemmas and frequently underwent multiple testing over many years. We identified 17 causal mutations and established the specific IBMFS diagnosis in 15 patients (18%) (table 3, see online supplementary figure S59).
Three of the successfully genotyped unclassified patients with IBMFS had predominantly neutropenia (see online supplementary table S4, patients 1–3). Two patients had mutations in known K/SCN genes: WAS and G6PC3. However, in one case the mutated gene, GATA2, was not known as a K/SCN gene,26–28 and biased testing for K/SCN-related genes would not have identified the causal genotype.
Nine of the successfully genotyped unclassified patients with IBMFS had multilineage cytopenia (see online supplementary table S4, patients 4–12). Five of these patients were ultimately diagnosed with DC due to mutations in telomere maintenance-associated genes (see online supplementary table S4, patients 4–8). The haematological phenotype of these patients with DC varied from predominantly anaemia to moderate aplastic anaemia and SAA. Interestingly, three other patients with pancytopenia were ultimately diagnosed with predominantly neutropenia syndrome (Warts–Hypogamaglobulinemia–Infection–Myelokathexis (see online supplementary table S4, patient 9), predominantly erythrocytopenia syndrome (DBA, see online supplementary table S4, patient 10) or predominantly thrombocytopenia syndrome (MYH9-related FT) (see online supplementary table S4, patient 11). These three patients would not have been genotyped if only pancytopenia-related genes had been sequenced.
Three genotyped unclassified patients with IBMFS belonged to a group of 10 patients who had SAA and no response to immunosuppressive therapy (see online supplementary table S4, patients 13–15). Two patients had mutations in pancytopenia-associated genes, RTEL1 and TERT. Surprisingly, one patient had a mutation in microtubule-associated serine/threonine kinase-like (MASTL), which would not have been normally tested in cases of SAA, as so far it has been associated only with FT.
Changing indications for implementation of cancer surveillance programme
In 20% of genotype cases, the results indicated a cancer surveillance programme and proper family counselling. One example is a patient with chronic severe neutropenia and marrow monosomy 7 (see online supplementary table S4, patient 1), who was found to have a mutation in GATA2. Another example is a patient with chronic moderate neutropenia (see online supplementary table S4, patient 2), solitary kidney and hypocellular bone marrow, who was found to have an activating WAS mutation.29
Expanding syndromes’ phenotype
Our study expanded the known phenotype of two syndromes. Neutropenia has not been reported as a feature of MYH9-associated FT.30 One of the patients with unclassified IBMFSs presented with early-onset thrombocytopenia and neutropenia, which persisted at moderate level (see online supplementary table S4, patient 11). The successful genotyping suggests that mutations in MYH9 may also cause neutropenia. The second syndrome is MASTL-associated disorder, which thus far has been linked only to non-syndromic FT, but the results in our study suggest that it can also be associated with SAA.
Cost consideration
We compared the cost of NGS assay to the cost of clinical testing for 30 patients with IBMFS enrolled at one of the CIMFR institutions (the Hospital for Sick Children, Toronto) between December 2010 and December 2013. Also, 21/30 had clinical genetic testing, averaged 5.95 tests/patient and US$4643/patient. These costs did not include expenses of DNA extraction and shipping to designated laboratories. The cost of NGS averaged $470/patient. This included reagents, sequencing service, bioinformatics and salary. In case of urgent testing without sequencing batching, the maximum price for NGS testing would be $2605/patient. This cost did not include a profit charge in the NGS cost, while this charge was incorporated in the service cost by the clinical laboratories. It is anticipated that rapid genotyping will not only reduce cost of genetic testing but also the cost of frequent clinic visits after presentation and other diagnostic ancillary laboratory tests such as skeletal survey, telomere length, adenosine deaminase levels, chromosome fragility test and pancreatic function tests.
Discussion
We evaluated the effectiveness of a new comprehensive NGS IBMFS gene panel assay on a large cohort of patients with IBMFSs and showed that it detects mutations with high sensitivity and precision. The test assisted in establishing a diagnosis in difficult cases and amended diagnoses that have been established solely on clinical basis. It is rapid, cost-effective and yields high-positive hits compared with the typical diagnostic odyssey that many of these patients with IBMFS encounter.
Our assay was effective in identifying causal mutations in 59% of the classified cases and led to amendment of clinical diagnoses in 9% of the genotyped cases. This exemplifies the potential pitfalls of targeting a specific diagnosis in patients with IBMFSs, even when the clinical features are highly suggestive of that disorder. These pitfalls can be overcome only by a comprehensive panel testing of all IBMFS genes that associated with single and/or multilineage cytopenias, and not by panel assays that are restricted to genes associated with one disease/phenotype.
The results of this study also underscore the advantage of comprehensive testing in unclassified IBMFSs. We genotyped and consequently established a diagnosis in 18% of these cases. Our study uncovered atypical presentations of patients with specific genotypes (eg, certain MASTL mutations in SAA) that allow expansion of clinical definitions of syndromes and refinement of their diagnostic criteria. Although the number of patients with SAA that did not respond to immunosuppressive therapy that tested herein was only 10, the proportion of successfully genotyped patients appears to be higher (30%) than that in previous reports (<5%).31–33 This is the first report of a patient with SAA who was tested and found positive for mutations in MASTL. MASTL-associated IBMFS is AD and is characterised by moderate thrombocytopenia. Platelets are of normal size and function. Haemoglobin levels and neutrophils were reported to be normal. Marrow megakaryocytes are typically reduced.34–36
Our study shows that the NGS assay can have a major impact on clinical care. For example, the amendment of clinical diagnosis of FA to DC in one of the patients indicates a need for substantially more intensive HSCT preparatory regimen to achieve successful engraftment. Further, patients with IBMFS with similar presentations were found to have syndromes that carry markedly different cancer risk. For examples, patients with isolated neutropenia were classified as having a GATA2-associated disorder (very high risk of leukaemia, no risk of carcinomas) or a CXCR4-associated disorder (low or potentially absence of a risk of leukaemia, risk of mucocutaneous carcinoma). Also, four patients with a clinical diagnosis of non-syndromic FT were accurately categorised as having either ANKRD26-associated FT and DC (substantial risk of MDS/AML) or MYH9-related FT (no risk of MDS/AML).
Not all patients were genotyped by the NGS assay. This might be due to incomplete target coverage (∼1%), exclusion of deep intronic areas, large indels, inability of bioinformatics tools to determine whether certain rare variant are causal and/or incomplete knowledge of all the IBMFS genes. Small indels and promoter mutations are captured by our panel design. In a proportion of the patients with FA, one allele of an early haematopoietic stem cells/progenitor undergoes spontaneous genetic correction and the respective developing precursors lose the increased chromosome fragility phenotype. This results in a mixture of healthy and FA cells and peripheral blood cell mosacism on chromosome fragility testing. In compound heterozygous cases, NGS will be able to detect the aberrant reads from non-corrected alleles. In most cases with homozygous mutations and mosacism, functional correction results in a sequence that is not identical to the wild-type one and the non-corrected mutant allele will still be identified by NGS. Genetic counselling should always be recommended, and the above limitations should be mentioned when results are disclosed to patients. Similar to reports of Sanger sequencing results, novel variants may be reported as likely positive or likely negative. Newly discovered genes can be incorporated in the panel as they are discovered by determining the precise coordinates of the fragments to be sequenced and designing oligonucleotides as described in online supplementary table S1. Hence, periodic repeat testing by updated gene panels may result in successful genotyping. Our assay can serve as a screening test before applying gene discovery methods such as exome sequencing.
In summary, our novel NGS IBMFS gene panel assay is a rapid, accurate and cost-effective strategy to genetically investigate patients with IBMFSs. The correct classification of IBMFs by NGS facilitates the more accurate medical management of these complex conditions. Therefore, we propose that NGS gene panels be considered as the first-line clinical molecular diagnostic test when the list of potentially mutated genes includes multiple candidates; this applies to the majority of patients with IBMFSs. Similar strategies may also be applied to other groups of genetic disorders with variable disease expression and presentation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors IG and HL contributed equally to the paper and should be considered co-first authors. IG performed research, analysed data and wrote the paper. HL performed research, analysed data and wrote the manuscript. BZ collected and analysed data. RK, CVF, RAY, JW, YP, MSi, JHL, JBr, BM, SA, MSt, RS, MB, VRB, LJ, LG and MC are study co-investigators, contributed vital data and review and/or edited the paper. LS is a study investigator, involved in study design, interpretation of results and edited the manuscript. SD and ER performed research, analysed data and interpreted results. AW evaluated and contributed clinical and genetic data. JBe is a study co-investigator, contributed analytical tools. PR and SM analysed data, interpreted results and wrote the manuscript. YD designed research, oversaw the project, analysed data and wrote the paper.
Funding This work was supported by grants from C17 Canadian Research Network and Candlelighters Canada, from the Nicola Kids’ Triathlon Fund and from the Canadian Institute of Health Research (funding reference 102528).
Competing interests None declared.
Ethics approval The Hospital for Sick Children Research Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.