Article Text

Abstract
Background Joubert syndrome (JS) is a recessive neurodevelopmental disorder characterised by hypotonia, ataxia, cognitive impairment, abnormal eye movements, respiratory control disturbances and a distinctive mid-hindbrain malformation. JS demonstrates substantial phenotypic variability and genetic heterogeneity. This study provides a comprehensive view of the current genetic basis, phenotypic range and gene–phenotype associations in JS.
Methods We sequenced 27 JS-associated genes in 440 affected individuals (375 families) from a cohort of 532 individuals (440 families) with JS, using molecular inversion probe-based targeted capture and next-generation sequencing. Variant pathogenicity was defined using the Combined Annotation Dependent Depletion algorithm with an optimised score cut-off.
Results We identified presumed causal variants in 62% of pedigrees, including the first B9D2 mutations associated with JS. 253 different mutations in 23 genes highlight the extreme genetic heterogeneity of JS. Phenotypic analysis revealed that only 34% of individuals have a ‘pure JS’ phenotype. Retinal disease is present in 30% of individuals, renal disease in 25%, coloboma in 17%, polydactyly in 15%, liver fibrosis in 14% and encephalocele in 8%. Loss of CEP290 function is associated with retinal dystrophy, while loss of TMEM67 function is associated with liver fibrosis and coloboma, but we observe no clear-cut distinction between JS subtypes.
Conclusions This work illustrates how combining advanced sequencing techniques with phenotypic data addresses extreme genetic heterogeneity to provide diagnostic and carrier testing, guide medical monitoring for progressive complications, facilitate interpretation of genome-wide sequencing results in individuals with a variety of phenotypes and enable gene-specific treatments in the future.
- Joubert syndrome
- ciliopathy
- genotype-phenotype
- next generation sequencing
- genetic heterogeneity
Statistics from Altmetric.com
Introduction
Joubert syndrome (JS, OMIM 213300) is a recessive neurodevelopmental disorder characterised by abnormal eye movements, respiratory control disturbances, cognitive impairment, hypotonia and ataxia.1–4 Diagnosis of JS relies on a pathognomonic combination of imaging findings on axial MRI: cerebellar vermis hypoplasia, thickened and horizontally oriented superior cerebellar peduncles and a deep interpeduncular fossa (the ‘Molar Tooth Sign’ (MTS)).5 In addition to these core central nervous system (CNS) features, subsets of individuals with JS have ocular (chorioretinal coloboma and progressive retinal dystrophy), kidney (nephronophthisis), liver (spectrum of ductal plate malformation and fibrosis) and/or skeletal (dystrophy and polydactyly) involvement. JS overlaps genetically and phenotypically with the more severe Meckel syndrome, often defined by co-occurrence of occipital encephalocele, cystic-dysplastic kidney disease, liver fibrosis, and perinatal lethality.6 Care of individuals with JS is complex, requiring surveillance for progressive complications and input from multiple medical subspecialists.
JS can be caused by recessive mutations in more than 27 genes, all of which encode proteins localising to the primary cilium or basal body.3 ,7 Primary cilia are microtubule-based organelles projecting from the surface of most differentiated cells where they serve as environmental sensors, transducing sensory, chemical or mechanical input, as well as signalling pathways (such as hedgehog) during development and homeostasis.8 Given the key role of this organelle in such a wide variety of processes, it is not surprising that its dysfunction leads to a number of human diseases collectively named ‘ciliopathies’.9 These disorders are unified not only by the underlying pathophysiology and shared genetic causes, but also by a wide array of overlapping phenotypes including cognitive dysfunction, CNS malformations, fibrocystic kidney disease, retinal degeneration, skeletal and craniofacial abnormalities, polydactyly and defects in left-right asymmetry.10
Ciliopathies, in general, and JS, in particular, display prominent genetic heterogeneity, that is, biallelic mutations in many different genes cause the same disorder, albeit with variable severity. Clinically, identifying the genetic causes and understanding gene–phenotype correlations are essential for providing diagnostic testing, prognostic information and treatment recommendations; however, until recently, it has not been possible to identify the genetic cause in the majority of affected individuals. The advent of next-generation sequencing has revolutionised the study of Mendelian disorders by accelerating novel gene discovery.11 Using JS as a paradigm, we highlight how next-generation sequencing combined with extensive phenotypic data can inform prognosis leading to improved medical monitoring in rare disorders, generate insights into the differential tolerance of genes to mutation and aid in interpreting genome-wide sequencing results in individuals with diverse phenotypes. Understanding the genetic architecture of Mendelian disorders is also leading to gene-specific treatments and improved patient care.
Methods
Subject ascertainment and phenotypic data
Participants were referred to the University of Washington (UW) Joubert Syndrome Research Program by the Joubert Syndrome and Related Disorders Foundation and clinical collaborators internationally (see Acknowledgements). All participants have clinical findings of JS (intellectual impairment, hypotonia, ataxia and/or oculomotor apraxia) and diagnostic or supportive brain imaging findings (MTS or cerebellar vermis hypoplasia), or they have a sibling with JS. Clinical data were obtained by direct examination of participants, review of medical records and structured questionnaires. Neurologically Normal Caucasian Control Panels (Coriell panels NDPT020 and NDPT090—http://ccr.coriell.org) were sequenced as controls.
Mutation identification
Using Molecular Inversion Probes (MIPs),12 all exons in genes associated with JS or the allelic disorder Meckel syndrome (AHI1, ARL13B, B9D1, B9D2, C2CD3, C5ORF42, CC2D2A, CEP290, CEP41, CSPP1, IFT172, INPP5E, KIF7, MKS1, NPHP1, OFD1, RPGRIP1L, TCTN1, TCTN2, TCTN3, TMEM138, TMEM216, TMEM231, TMEM237, TMEM67, TTC12B and ZNF423;13–36 details in online supplementary table S1) were captured using 100 ng of genomic DNA isolated from blood or saliva. Captured DNA was PCR amplified and sequenced on either the Illumina HiSeq or MiSeq platform. Sequence reads were mapped using the Burrows-Wheeler Aligner (V.0.5.9). Variants were called using the Genome Analysis Toolkit (V.2.5–2) and annotated with SeattleSeq (http://snp.gs.washington.edu/SeattleSeqAnnotation138/). We also included data previously generated by Sanger sequencing of individual genes in subsets of samples. We used the Combined Annotation Dependent Depletion (CADD) algorithm to estimate the deleteriousness of variants (V.1.1),37 and considered all nonsense, frameshift and canonical splice-site mutations to be deleterious, regardless of CADD score. We defined a cause as the presence of ≥2 rare deleterious variants (RDVs) or a homozygous RDV in one gene in an affected individual. RDVs that were of high quality (depth ≥25, quality by depth >5 and heterozygous allele balance <0.8) were not confirmed by Sanger sequencing based on the previously demonstrated high sensitivity and specificity of the MIPs method for well-covered variants12; however, in affected individuals with one high-quality RDV, we did perform Sanger sequencing to confirm second RDVs that did not meet the above-mentioned quality criteria.
Statistical analysis
We tested the significance of associations between clinical features, as well as between features and genetic causes, using the χ2 or Fisher's exact tests (SAS, V.9.4; SAS Institute, Cary, North Carolina, USA). We present ORs and 95% CIs as measures of these correlations. The Bonferroni method was used to correct for multiple hypothesis testing.
Results
UW JS cohort
The study cohort comprised 532 affected participants from 440 families, 79 families having >1 affected individual. Participants were recruited from 29 countries, the majority (59%) residing in North America. Nineteen per cent of the families reported consanguinity. The mean age of the affected participants at the time of the analysis was 13.1 years (SD 9.1), with 34% of individuals <10 years of age and 30% 10–20 years of age. Fifty-six per cent were male (table 1). The large size of the cohort and worldwide ascertainment based on brain imaging and neurological findings provide a relatively unbiased spectrum of the disorder.
Demographic characteristics of the University of Washington Joubert syndrome cohort
Multiorgan involvement is common and the ‘pure JS’ phenotype occurs in a minority of individuals
In addition to the core diagnostic features for JS (MTS, hypotonia, ataxia, cognitive dysfunction, abnormal breathing pattern and oculomotor apraxia) that were part of the inclusion criteria, several extra-CNS features are commonly described in JS. Based on the presence of these features, various subtypes of JS have been proposed:2 ‘pure’ JS (core diagnostic features only), JS plus retinal dystrophy, JS plus cystic kidney disease, JS plus retinal–renal involvement, JS plus liver fibrosis and JS plus oral-facial-digital features. Therefore, we systematically assessed the relevant features (see online supplementary table S2) in the cohort. As a consequence of the worldwide recruitment required to collect a large cohort for a rare disorder, the ascertainment of clinical features was variable. To be conservative in calculating the prevalence of each feature, we restricted our analysis to individuals for whom definite positive or negative information was available for a given feature; consequently, the denominator for calculating the frequency of individual features varies accordingly. Retinal dystrophy (n=99/329, 30%) and renal disease (n=102/407, 25%) were the most common associated features, followed by coloboma (n=56/330, 17%), polydactyly (n=56/387, 15%), liver fibrosis (n=50/362, 14%) and encephalocele (n=29/386, 8%) (figure 1A). When considering only the individuals for whom definite information was available for all six associated features (n=201), only 68 (33.8%) had the ‘pure JS’ phenotype (see online supplementary table S3).

Phenotypic analysis of a large Joubert syndrome (JS) cohort. (A) Bar graph indicating the prevalence of major associated features. Absolute numbers are indicated below each bar and 95% CIs are presented. Information about each feature was not available in every subject, so the denominators are different for each variable. (B) ORs for the association between pairs of features. Hepatic disease and coloboma are highly associated with each other while encephalocele and polydactyly, retinal and renal disease, and hepatic and renal disease are less strongly associated with each other. Precise ORs with 95% CIs are indicated for the four statistically significant (***) associations. Detailed ORs and CIs for all pairwise possible associations are presented in online supplementary table S4.
We next evaluated whether any of the major features were associated with each other. Liver fibrosis and coloboma were strongly associated (OR 6.5; 95% CI 3.2–13.4), that is, the likelihood of having liver fibrosis in individuals with coloboma was 6.5 times the likelihood of having liver fibrosis in individuals without coloboma. Retinal dystrophy and kidney disease (OR 3.0; 95% CI 1.7 to 5.2), liver fibrosis and kidney disease (OR 3.0; 95% CI 1.6 to 5.5) and polydactyly and encephalocele (OR 2.8; 95% CI 1.03 to 7.8) were more weakly associated with each other (figure 1B and see online supplementary table S4). In addition, we observed multiple combinations of features in subsets of individuals, often precluding categorisation into one of the proposed subtypes (see online supplementary table S3). For example, individuals presenting with the combination of liver fibrosis and kidney disease could be categorised as either ‘JS plus kidney disease’ or ‘JS plus liver disease’. While the most frequent associations of features are consistent with the proposed JS subtypes, the broad range of additional combinations observed indicates that no clear-cut distinction exists between subtypes.
Multiple additional clinical features
A variety of other clinically important features were documented in medical records and by families but were not systematically queried across the entire cohort (table 2). Additional brain abnormalities were identified in 91 individuals, most commonly ventriculomegaly, and more rarely heterotopia, agenesis of the corpus callosum and polymicrogyria. This is likely an under-ascertainment compared with prior studies38 since a detailed review of the brain imaging studies was not part of this study. Additional eye findings were also commonly reported in our cohort, including strabismus and ptosis in 167 and 104 individuals, respectively. Seizures were described in 55 individuals. Other, less common, features included scoliosis (n=28), cleft palate (n=20), hearing loss (n=16), tongue tumours (n=17), oral frenulae (n=9), heart defects (n=7) and a variety of mental health problems such as anxiety, aggression, depression and autism (total n=47). Since these features were not systematically assessed across the cohort, only minimum prevalence estimates can be calculated.
Additional features observed in individuals with Joubert syndrome
Comprehensive sequencing identifies the presumed genetic cause in 62% of JS families
We sequenced 27 JS-associated genes in 428 affected individuals from 363 families for whom DNA was available using MIP-targeted capture followed by next-generation sequencing. We previously demonstrated, using a subset of this cohort, that this method has 99.5% sensitivity and 98% positive predictive value for variant detection at covered basepairs compared with Sanger sequencing.12 The MIP target included all coding positions and neighbouring intronic basepairs (see online supplementary table S1), and >89% of basepairs were adequately covered (≥8X) for all genes except INPP5E (75% covered) (see online supplementary figure S1). We also included previous Sanger sequencing data, as well as sequencing data from clinical testing when available (n=12), bringing the total number of affected individuals with sequencing data to 440 from 375 families. Based on the estimated prevalence of JS (∼1/80 000 Northern Europeans3) and the genetic heterogeneity of the disease, we excluded variants with a minor allele frequency (MAF) >0.2% in the Exome Variant Server (http://evs.gs.washington.edu/EVS/). We considered all nonsense, frameshift and canonical splice-site mutations to be deleterious. We assessed the predicted deleteriousness of missense, synonymous and intronic variants using the CADD score algorithm,37 which considers multiple available prediction techniques including conservation across species and protein function, and has the advantage of providing a score for all possible variants on a single scale. We selected the CADD score cut-off (11) for defining RDVs by maximising the number of affected individuals with genes harbouring two rare variants (or a homozygous rare variant), while minimising the number of controls with genes harbouring similar variants, an approach akin to generating a receiver operating characteristic curve (see online supplementary figure S2). For missense variants, using the CADD score identified more presumed causes in the JS cohort compared with Polyphen2 without increasing the false positive rate in controls (data not shown).
We defined a cause as the presence of ≥2 RDVs (or a homozygous RDV) in one gene in an affected individual. Using this definition and all available sequencing data, we identified the presumed genetic cause in 279 individuals from 232/375 families (62%) overall (figure 2), 77% in consanguineous families and 76% in families with >1 affected individual. The higher rate in the consanguineous families is likely due to the higher probability of calling a single homozygous variant compared with the probability of calling two different heterozygous variants in the non-consanguineous families. Similarly, in 9% of families for whom we were able to sequence >1 affected individual, we initially identified two RDVs in only one of the affected individuals. This likely accounts for the higher solve rate in multiplex families compared with families with only one affected child. In contrast to the results in affected individuals, 5/182 unrelated control individuals carried ≥2 RDVs in one of the known genes (see online supplementary table S5). In 68/70 (97%) families for which parental DNA was available, we confirmed that the identified compound heterozygous RDVs are in trans, excluding two samples from further analysis. We did not sequence parents of children with homozygous or hemizygous RDVs (90 families). Parental samples were not available for controls.
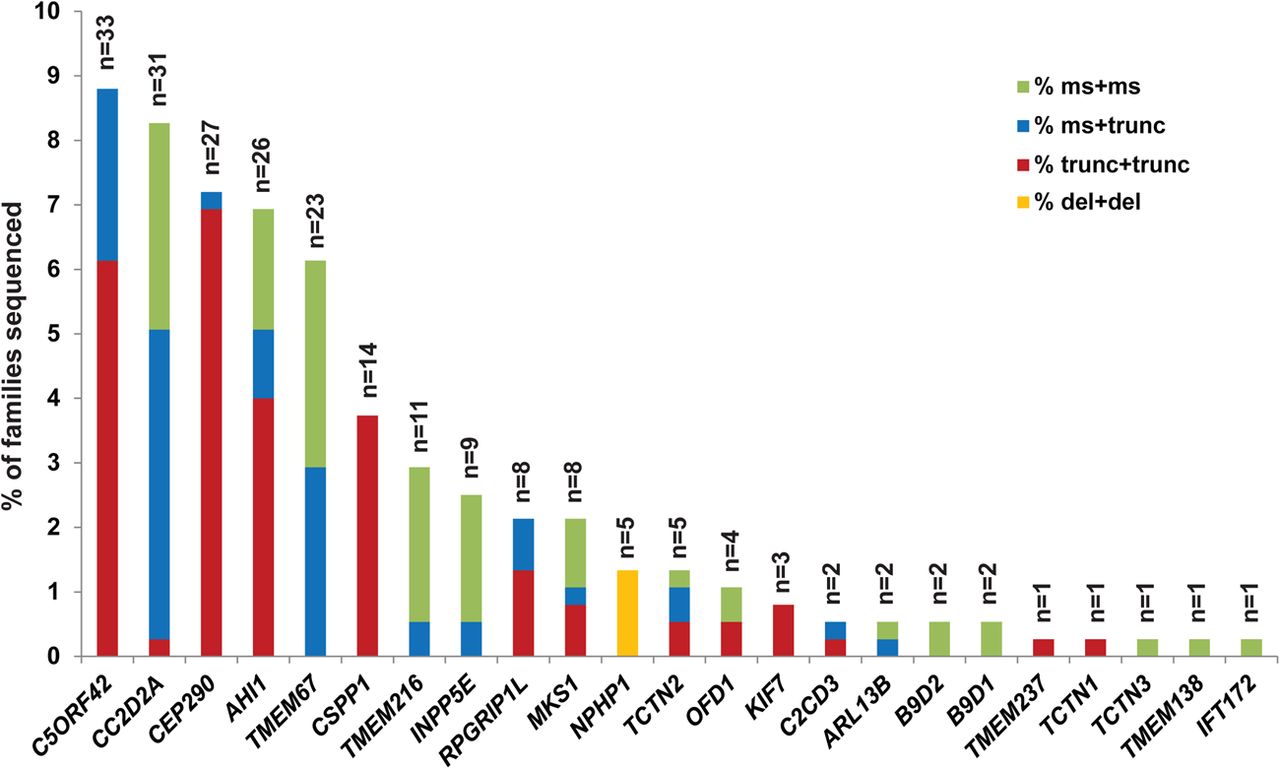
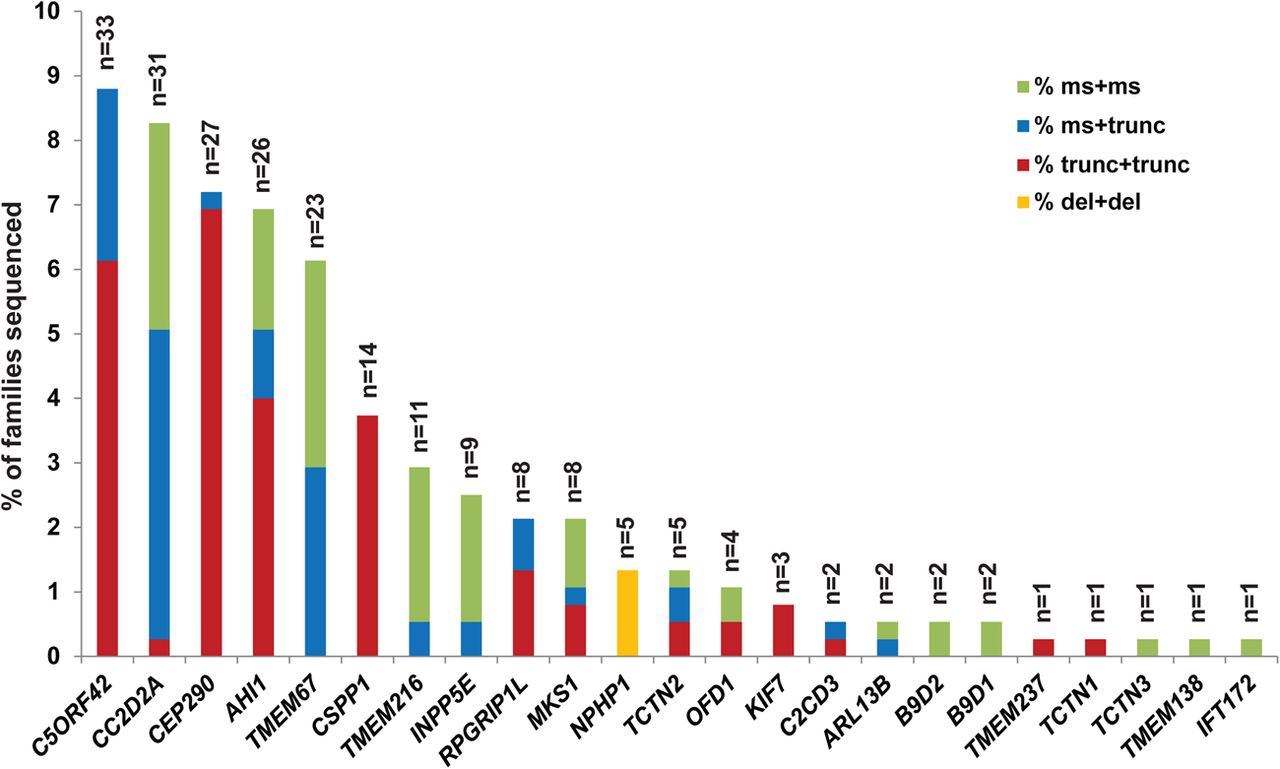
Genetic causes in a large Joubert syndrome cohort. Bar graph indicating the proportion of individuals with JS carrying two rare deleterious variants in each gene. Each bar is broken down to illustrate the relative frequency of the observed mutations in each gene: red indicates two truncating mutations (including nonsense, frameshift and canonical splice-site mutations), blue indicates one truncating and one missense mutation (including small in-frame indels), green indicates two missense mutations or small in-frame indels and orange represents larger deletions.
Despite satisfying our criteria (MAF<0.2%, CADD>11), the variants in 12 families did not meet the American College of Medical Genetics and Genomics variant interpretation categories 1, 2 or 3.39 In 8 of these 12 families, one of the RDVs is a splice variant beyond±2 basepairs from the intron–exon junction, for which the functional effect on splicing has not been assessed. In 4/12 families, one RDV is a synonymous variant whose functional effect has not been evaluated. Therefore, we list these families separately in online supplementary table S5 and excluded them from gene–phenotype analyses.
In addition, we identified five families with pairs of RDVs in each of two genes (see online supplementary table S6). In 3/5, the variants in one gene appeared much more likely to be causal than the variants in the second gene (eg, a homozygous frameshift mutation in C5ORF42 vs two missense variants in CSPP1, which harbours exclusively truncating mutations in our cohort). In these three families, the more likely cause was retained for the subsequent analyses. In the other two families, we could not determine the cause and excluded them from the subsequent genetic analyses. Of note, based on the clinical information available, the phenotypic severity in these five individuals was not substantially different from the rest of the cohort.
Including only the families with conservatively called genetic causes, five genes (C5ORF42, CC2D2A, AHI1, CEP290 and TMEM67) each account for JS in ∼6–9% of JS, three genes (CSPP1, TMEM216 and INPP5E) for ∼3% each and six genes for ∼1–2%, while the remaining nine genes each account for JS in only 1–2 families. We also identified B9D2 mutations as the genetic cause in two families, further extending the known genetic overlap between JS and the more severe Meckel syndrome. A detailed phenotypic description of the two individuals with B9D2 mutations is presented in online supplementary table S7. CEP41, TMEM138, TMEM231 and ZNF423 do not harbour ≥2 or homozygous RDVs in any affected individuals. A single affected individual carries one synonymous and one missense variant in TTC21B; however, this individual also carries a homozygous nonsense variant in C2CD3 that is predicted to truncate the protein near the N-terminus (see online supplementary table S6).
Further examination of the sequence data revealed variation in the types of mutations across the different genes. Considering all nonsense, frameshift and canonical splice-site mutations as truncating, we observed that CEP290, CSPP1 and C5ORF42 mostly harbour a combination of two truncating mutations, CC2D2A and TMEM67 tend to have ≥1 missense mutation, and TMEM216 and INPP5E have mainly two missense mutations. All individuals with JS caused by mutations in NPHP1 (n=5) harbour the previously described deletion24 in a homozygous state, and no causal point mutations were identified in this gene. The differences in mutation types across the genes were statistically significant (see online supplementary figure S3).
While the majority of RDVs were unique, we identified a subset of RDVs present in ≥3 families not known to be related (see online supplementary table S8). TMEM216 R73L is common in families of Ashkenazi Jewish descent,34 and accounts for most of the families with TMEM216 mutations. Two C5ORF42 RDVs (p.Gly2663Alafs*40 and W2593*) were found homozygous in six families of Saudi Arabian descent. The p.Gly2663Alafs*40 variant has been previously associated with both JS and Meckel syndrome in Saudi Arabian families.40 ,41 One CC2D2A RDV (P1122S) was found homozygous in three families of Saudi Arabian descent. In three unrelated Brazilian families, the same combination of two CSPP1 RDVs was identified, suggesting that they might in fact be related.20 None of the other recurring RDVs appeared to be associated with specific ethnic groups, so they may represent mutation hotspots (such as CEP290 G1890* identified in 10 unrelated families from 3 continents).
Gene–phenotype correlations
We next examined associations between the non-CNS features of JS and each genetic cause (figure 3, see online supplementary table S9) and observed several significant gene–phenotype correlations: CEP290 mutations with retinal dystrophy (OR 22.9, 95% CI 6.7 to 78.4; p<0.0001) and cystic kidney disease (OR 3.3, 95% CI 1.6 to 7.1; p=0.001); TMEM67 with liver fibrosis (OR 17.3, 95% CI 7.2 to 42.0; p<0.0001) and coloboma (OR 22.9, 95% CI 8.6 to 61.1; p<0.0001); C5ORF42 with polydactyly (OR 2.7, 95% CI 1.2 to 5.9; p=0.01); OFD1 with encephalocele (OR 13.1, 95% CI 1.8 to 97.0; p=0.03); TCTN2 with encephalocele (OR 13.6, 95% CI 2.6 to 70.8; p=0.007) and polydactyly (OR 18.7, 95% CI 1.9 to 182.9; p=0.01). Even after Bonferroni correction for multiple hypothesis testing, the associations between TMEM67 and liver disease and coloboma, and that between CEP290 and retinal dystrophy remained statistically significant (p<0.0001). In addition, a negative correlation was observed between TMEM67 mutations and retinal disease (OR 0.1, 95% CI 0.01 to 0.8; p=0.006), indicating that individuals with TMEM67 mutations are less likely to be diagnosed with retinal disease than those without mutations in this gene. When counselling families, the absolute prevalence of clinical features may be more useful than ORs, so this information is provided in online supplementary figure S4.

{kind=link}
{kind=link}
{kind=link}
Gene–phenotype correlation in Joubert syndrome. Bar graph indicating for each of the more frequently involved genes, and for two genes with significant phenotypic associations, the OR for each of the six commonly associated features: retinal disease, renal disease, hepatic disease, coloboma, polydactyly and encephalocele. Statistically significant ORs (Fisher's exact test or χ2 test) are marked with an asterisk (***). CIs are omitted for clarity but are listed in online supplementary table S9.
Although we cannot test the statistical significance of genetic associations with non-systematically assessed clinical features, several possible associations are notable. Both individuals with C2CD3 mutations had oral features including oral frenulae and/or cleft palate, suggesting C2CD3 mutations may lead to an OFD-like phenotype.2 However, among the individuals with oral features (n=46), the majority did not have mutations in C2CD3 (or OFD1). Likewise, two of three individuals with KIF7 mutations had agenesis of the corpus callosum (while the status of the corpus callosum in the third individual was unknown), consistent with a KIF7-related ‘acro-callosal’ subtype of JS. Again, however, the majority of individuals with agenesis of the corpus callosum (n=14) had mutations in other genes without a clear predominance of one genetic cause. None of the 55 individuals with seizures had causal CEP290 mutations, despite CEP290 loss of function being the third most common cause of JS, suggesting a negative association.
Discussion
Presumed genetic cause of JS identified in 62% of families
Just over 10 years ago the first genetic causes of JS were identified.13 ,24 Now, we can determine the presumed genetic cause in 62% of individuals with JS using the highly efficient MIP capture technique, next-generation sequencing and an optimised CADD score cut-off to identify causal variants in 27 JS/Meckel genes. Five genes (C5ORF42, CC2D2A, CEP290, AHI1 and TMEM67) account for the majority of affected individuals, while nine genes are mutated in <15 families, and nine more genes are mutated in only 1–2 families. In two families with JS, we identified causal mutations in the Meckel-associated gene B9D2, further expanding the allelism between JS and Meckel syndrome. Not surprisingly, B9D2 is part of a transition zone subcomplex (with MKS1 and B9D1) that regulates protein trafficking in and out of the cilium.42
These findings illustrate the extreme genetic heterogeneity of JS. Therefore, given that no single gene predominates as a cause for JS, the most efficient method for clinical diagnostic testing is next-generation sequencing of all known JS genes through targeted gene panels or whole-exome sequencing. The advantage of the MIP capture technique lies in its low cost and flexibility, allowing easy addition of newly identified JS genes to the target. For laboratories without a specific interest in JS, whole-exome sequencing might be more practical since it does not require any specialised set-up.
The genetic cause remains unidentified in 38% of families in our cohort. This may be due to mutations in genes not yet associated with JS, or variants in the known genes that were missed by our current techniques, either because they are inadequately covered in our data, located in non-coding regions, not called using our analysis pipeline, or not recognised as deleterious. Given the high coverage obtained for all but one gene (INPP5E) and the efficiency of MIP capture for identifying variants in the target regions,12 it is likely that a sizeable fraction of the missed variants lie in non-coding regions that affect gene expression level, splicing or translation. Identifying these variants and understanding their significance will require integrating data from variant rating algorithms like CADD, global assessments of chromatin structure and regulatory elements from projects such as ENCODE43 and targeted functional assays in affected cell lines, animal models or in vitro systems.
Clinical utility of gene–phenotype correlations and phenotypic associations
Gene–phenotype correlations in well-characterised, comprehensively sequenced cohorts translate directly into improved prognostic information and medical management for individuals with JS. For instance, results from this study indicate that individuals with JS harbouring causal mutations in TMEM67 have a higher risk of developing liver fibrosis, necessitating closer monitoring to allow early diagnosis and treatment of portal hypertension. Likewise, individuals with causal mutations in CEP290 require closer surveillance for retinal dystrophy. Our findings validate prior results from smaller cohorts focused on single genes44 ,45 ,46 and also identify additional positive and negative correlations. For example, individuals with causal mutations in TMEM67 appear less likely to develop retinal disease and may require less frequent monitoring for this complication. Even when the genetic cause is unknown, phenotypic associations can also guide management and surveillance; for example, individuals with JS and retinal dystrophy should be monitored more closely for renal dysfunction, and those with coloboma should be monitored more closely for liver fibrosis.
While the strongest phenotypic associations observed in this cohort are consistent with previously described JS-subtypes such as COACH syndrome,45 ,46 and the retinal-renal form of JS,44 we did not observe clear-cut distinctions between phenotypic subgroups corresponding to specific genetic causes. The MTS provides a unifying feature for all affected individuals in our cohort, but the distribution of associated phenotypes highlights the phenotypic variability and overlap with other ciliopathies. This is particularly well illustrated by the individuals with mutations in the OFD-associated genes C2CD3 or OFD1 who have oral features, consistent with an OFD-like JS subtype; however, most individuals with oral features in our cohort harbour mutations in other genes. Therefore, phenotypic subtyping is of limited clinical value for guiding molecular genetic testing. Fortuitously, next-generation sequencing panels now preclude the need for prioritising single gene tests. Nonetheless, grouping individuals by genetic cause or clinical phenotype retains value for determining their risk of developing progressive features and guiding clinical management as described above.
Gene-specific mutation patterns provide insights into gene function
The observed gene–phenotype correlations, along with the gene-specific mutation distributions, provide information about the function of the different genes. Genes associated preferentially with particular phenotypes suggest a specific or more important role for these genes in the affected organ systems. For instance, the association of CEP290 mutations with retinal dystrophy in JS and Leber congenital amaurosis44 ,47 confirms the importance of CEP290 function in the human retina, as seen in animal models.
The distribution of mutation types harboured by each gene also reveals information about gene function. For instance, the near-absence of biallelic truncating mutations in some genes suggests that full loss of function for these genes is poorly tolerated in humans, leading to more severe phenotypes, such as Meckel syndrome or early fetal lethality. In support of this hypothesis, fetuses with Meckel syndrome tend to carry two truncating mutations in CC2D2A and TMEM67 compared with individuals with JS who usually carry at least one missense mutation as previously described.48–50 Likewise, biallelic truncating mutations in TMEM216 and INPP5E have not been previously identified in individuals with JS and are not found in our cohort.22 ,33 ,34 ,51 In contrast, virtually all individuals with JS due to mutations in CSPP1 or CEP290 harbour two truncating variants in these genes, indicating that severe loss of function is required to cause JS. This type of gene-specific information should be considered when interpreting the significance of newly identified sequence variants, in combination with allele frequency in controls, deleteriousness prediction algorithms and the phenotype of the affected individual. For example, missense mutations in CEP290 or CSPP1 detected by targeted or genome-wide clinical sequencing are less likely to be clinically significant than missense mutations in TMEM216 or INPP5E. A further consequence of the gene-specific distribution of mutation types lies in the development of potential specific therapies: genes harbouring a majority of nonsense mutations such as CEP290 may be amenable to read-through therapies,52 while this therapeutic direction would be less valuable for genes harbouring mainly missense mutations.
Limitations
While larger than previously published studies, our analysis is still limited by the small number of individuals with two RDVs in several genes associated with JS, precluding statistically significant gene–phenotype correlations for these genetic causes. This is an inherent limitation to the study of rare disorders with prominent genetic heterogeneity. Similarly, the relative rarity of JS necessitates the worldwide enrolment of study participants; consequently, phenotypic assessment is inhomogeneous and some features, especially neurodevelopmental outcome, are difficult to assess at a distance. This is currently a universal problem in the field of rare disorder genetics, where, for the first time, genetic data are more easily available than phenotypic data. In this study, we made every effort to use conservative assumptions for tests of statistical significance; however, until validated by other studies, these results should be translated into clinical practice with caution.
Impact of next-generation sequencing on diagnosis and treatment of Mendelian disorders
In summary, this work illustrates how applying advanced DNA sequencing technologies and improved functional prediction algorithms to large, well-characterised cohorts is enhancing our understanding of the genetic architecture and gene–phenotype correlations in rare Mendelian disorders. Identifying the genetic cause empowers individuals with JS and their families to make family planning decisions, and gene–phenotype correlations provide more reliable prognostic information leading to individually tailored, organ-specific surveillance, thereby improving the health and longevity of affected individuals while conserving healthcare costs. In parallel, identifying the genetic causes of Mendelian disorders is required for developing and applying gene-specific treatments. Similar to recent breakthroughs in cancer treatment based on genomic information (reviewed in Sameek and Chinnaiyan),53 understanding the genetic causes of Mendelian disorders will inform future gene-specific treatments and is a major step towards personalised medicine for affected individuals.
Acknowledgments
We greatly appreciate the participation of all of the individuals with JS and their families. We also thank the Joubert Syndrome and Related Disorders Foundation and the innumerable clinicians who have referred participants to us over many years, including Jumana Al-Aama, Beth Allen, Ala'a Arafat, Mutluay Arslan, Louis Bartoshesky, Erawati Bawle, Anastashia Boli, Carsten Bönnemann, Sarah Bowdin, Steven Braddock, John Carey, Umran Cetincelik, Alicia Chan, David Chitayat, Juan Chemke, Krystyna Chrzanowska, Brian Chung, Robin Clark, Vida Culic, Duygu Cura, Cynthia Curry, Sumita Danda, Stephen Deputy, Charu Deshpande, William Dobyns, Emily Doherty, Lisa Ewans, Michael Gabbett, Özlem Giray, Himanshu Goel, Elaine Goh, Crystal Grimsley, Sachin Gupta, Jonathan Holt, Niels Illum, Olafur Indridason, A. Micheil Innes, Louise Izatt, Sujatha Jagadeesh, Usha Kini, Jarkko Kirjavainen, Edwin Kirk, Antonie Kline, Kristleifur Kristjannson, Mark Labinksy, Yves Lacassie, Amparo Lopez Lafuente, James Lemons, Richard Leventer, Jan Liebeit, Shawn Lipinski, Sally Lynch, Alan Ma, Richard Macias, Sahar Mansour, Bernard Maria, Isabelle Maystadt, Carole McKeown, Scott McLean, Nancy Mendelsohn, Markella Mikkelsen, Vinod Misra, Sheela Nampoothiri, Vinodh Narayanan, David Neubauer, Ann Olney, Wendy Osterling, Alex Paciorkowski, Shashidhar Pai, Ivan Pavkovic, Joan Pellegrino, Ruthann Pfau, Joseph Pinter, Kate Pope, Gerald Raymond, Miriam Regev, James Reggin, Janet Rennie, Anne Ronan, Alan Shanske, Lisa Sharf, Lori Skallerud, Diana Smith, Rhonda Spiro, Zornitza Stark, Lois Starr, Helen Stewart, Anne Summers, Krzysztof Szczaluba, David Tilstra, John Tolmie, Priya Verghese, Alain Verloes, Julie Vogt, Richard Webster, Kara Weisiger, Mark Wells, Kevin White, Sue White, Dorota Wicher, Robert Wildin, Denise Williams, Meredith Wilson, Barry Wolf, Lisa Worgan, Grace Yoon, Takehito Yokoi, Marc Yudkoff, Elaine Zackai. We thank Karen Barnett for help establishing the cohort, Elizabeth Blue for comments on the manuscript, and Stephan Neuhauss and Anita Rauch for their support for R.B.-G.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors RB-G, JCD, IGP, PC, MAP, IG, JS, and DD participated in the design of the study. JCD, IGP, BJO, DMK, GEI, CRI, NG, JA, EAB, DO, AA, RRA, LL, CL, LM, AG-C, HO, GH, BT, MT, MAP, UWCMG and DD collected and/or generated the data. RB-G, JCD, IGP, BJO, TCR, EAB, NdL, UWCMG, and DD analysed and interpreted the data. RBG, JCD, IGP and DD drafted the manuscript. All coauthors read and approved the final manuscript.
Funding RB-G was supported by a Swiss NSF grant Ambizione-SCORE PZ00P3_142404/1. The following authors received support from the National Institutes of Health: M.A.P. K23NS45832, I.A.G. K24HD046712, D.D R01NS064077. D.D. also received funding from a March of Dimes Basil O'Connor Starter Scholar Research Award, The Arc of Washington Trust Fund, and private donations from families of children with Joubert syndrome. The work was also supported by the University of Washington Intellectual and Developmental Disabilities Research Center Genetics Core (National Institutes of Health U54HD083091). Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 1U54HG006493 to Drs Debbie Nickerson, JS and Michael Bamshad.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Institutional Review Boards at University of Washington and Seattle Children’s Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data are included in the manuscript and online supplementary data.