Article Text

Abstract
Background Mutations of SCN8A encoding the neuronal voltage-gated sodium channel NaV1.6 are associated with early-infantile epileptic encephalopathy type 13 (EIEE13) and intellectual disability. Using clinical exome sequencing, we have detected three novel de novo SCN8A mutations in patients with intellectual disabilities, and variable clinical features including seizures in two patients. To determine the causality of these SCN8A mutations in the disease of those three patients, we aimed to study the (dys)function of the mutant sodium channels.
Methods The functional consequences of the three SCN8A mutations were assessed using electrophysiological analyses in transfected cells. Genotype–phenotype correlations of these and other cases were related to the functional analyses.
Results The first mutant displayed a 10 mV hyperpolarising shift in voltage dependence of activation (gain of function), the second did not form functional channels (loss of function), while the third mutation was functionally indistinguishable from the wildtype channel.
Conclusions Comparison of the clinical features of these patients with those in the literature suggests that gain-of-function mutations are associated with severe EIEE, while heterozygous loss-of-function mutations cause intellectual disability with or without seizures. These data demonstrate that functional analysis of missense mutations detected by clinical exome sequencing, both inherited and de novo, is valuable for clinical interpretation in the age of massive parallel sequencing.
- Epilepsy and seizures
- Movement disorders (other than Parkinsons)
- intelectual disability
- sodium channel
- encephalopathy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Epilepsy and seizures
- Movement disorders (other than Parkinsons)
- intelectual disability
- sodium channel
- encephalopathy
Introduction
Exome sequencing has revolutionised the detection of disease-causing mutations, both in research and in diagnostic laboratories. The ease of exome-wide sequencing has led to the discovery of unanticipated associations between gene mutations and disease. However, there is a risk of overinterpreting the role of private mutations in a given gene with disease. Thus, a combination of segregation analysis, metabolic testing and/or functional studies is increasingly important to establish the link between isolated mutations and a patient's disorder.
Nine genes encoding distinct voltage-gated Na+ channel α subunits NaV1.1–1.9 have been identified and functionally characterised.1 The gene SCN8A encodes the NaV1.6 α subunit, which is abundantly expressed throughout the central nervous system, including high expression in the cerebellum, hippocampus, cortex and olfactory bulb.2–6 Primary motor neurons as well as cerebellar Purkinje and granule cells have strong expression of NaV1.6, suggesting a role in motor control.2 ,3 ,7 NaV1.6 channels are enriched at the neuronal axon initial segment and nodes of Ranvier, where they promote neuronal excitability by participating in the initiation and propagation of action potentials.8–12 NaV1.6 channels generate persistent current, hyperpolarised thresholds of activation compared with other NaV channels, and resurgent current (reviewed in13). These biophysical properties make NaV1.6 a critical mediator of neuronal excitability.
Homozygous null mice lacking functional NaV1.6 display severe motor deficits4 ,14–16 and impaired learning,17 while heterozygous null animals display absence epilepsy,18 disturbed sleep pattern19 and anxiety.13 ,20 Pathological-induced or drug-induced increases in NaV1.6 activity have been associated with susceptibility to epilepsy.21–23 In humans, an inherited frameshift mutation in SCN8A was identified in a child with intellectual disability (ID), ataxia and attention-deficit hyperactivity disorder (ADHD).24 This frameshift mutation was also present in the patient's mother and maternal aunt, who had ID without ataxia, and in a maternal nephew with isolated ADHD. In 2012, the first de novo SCN8A missense mutation was described in a child with severe epileptic encephalopathy.25 Recently, >30 de novo mutations in SCN8A have been identified in patients suffering from early-infantile epileptic encephalopathy type 13 (EIEE13), characterised by early-onset (refractory) epilepsy, features of autism and ID.26–32 Only three of these mutations have been subjected to functional analysis to determine the consequences for NaV1.6 channel activity.25 ,28 ,32 Two of the tested mutants, p.(Asn1768Asp) and p.(Thr767Ile), induced gain-of-function effects and hyperactivity, suggesting that EIEE13 can be caused by elevated NaV1.6 activity.
Here, using clinical exome sequencing in patients with ID, we identified three novel de novo mutations in SCN8A. The patients had ID with additional variable symptoms, including seizures, abnormalities on brain MRI, spasticity and facial dysmorphisms. We carried out electrophysiological analysis to characterise the effects of the identified mutations on the activity of the mutant NaV1.6 channels. This analysis demonstrated one mutation with gain-of-function and elevated channel activity, one with apparent loss of function and a third with no detectable functional effects. These results are discussed with respect to the individual clinical phenotypes.
Methods
Patients and clinical exome sequencing
The patients were referred to the Department of Human Genetics of the Radboud University Medical Center (Nijmegen, the Netherlands) for genetic diagnostic evaluation of unexplained ID/developmental delay and/or a movement disorder. Clinical exome sequencing was part of the routine diagnostic procedure. Previous genome-wide chromosomal analyses gave normal results. Patients 1 and 3 were assessed through a family-based exome sequencing (‘trio’-exome sequencing) approach.33 ,34 Patient 2, who also had ID, was part of a cohort of approximately 100 patients with movement disorders, predominantly ataxia or spastic paraplegia. Exome sequencing was performed essentially as described previously35 and in online supplementary data.
Cell culture and transfection
Human embryonic kidney cells (HEK293) were grown at 37°C in Dulbecco's modified Eagle's medium (Biowhittaker Europe, Vervier, Belgium) supplemented with 10% (v/v) fetal calf serum (PAA Laboratories, Linz, Austria), non-essential amino acids and 2 mM L-glutamine in a humidified 5% (v/v) CO2 atmosphere. Cells were seeded in 12-well plates and subsequently transfected with 1.5 μg of NaV1.6 construct and 150 ng enhanced green fluorescent protein using Lipofectamine 2000 (Invitrogen). Three mutations were introduced into the tetrodotoxin (TTX)-resistant derivative of the murine NaV1.6 cDNA clone NaV1.6R as described previously.25 ,28 ,32 Analysis of human mutations in the context of this TTX-resistant cDNA has facilitated the delineation of biophysical changes produced by human mutations in Nav1.6.25 ,28 The three substituted amino acids (D58, N984 and G1451) are conserved between human and mouse NaV1.6 (figure 1). The 6 kb open-reading frame was sequenced to confirm the absence of additional mutations. The day after transfection, cells were seeded on glass coverslips coated with 50 µg/mL of fibronectin (Roche, Mannheim, Germany). Approximately 20 h after transfection, cells were placed in the recording chamber and selected for recording based on intensity of fluorescent reporter.
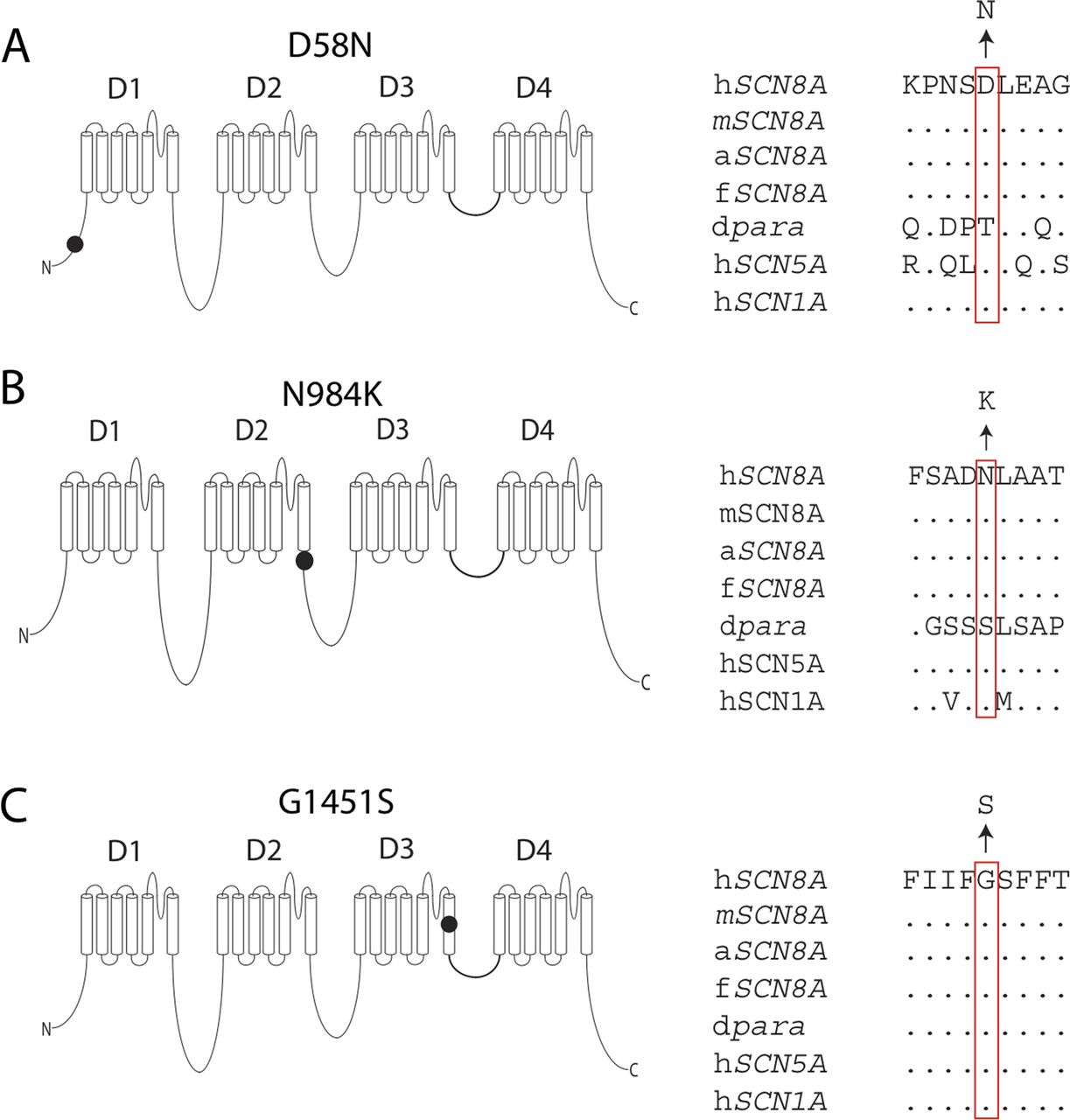
Localisation and evolutionary conservation of three newly identified de novo mutations in SCN8A encoding the sodium channel NaV1.6. (A) The D58 residue in the N-terminus. (B) N984 in the intracellular loop close to the sixth transmembrane helix of domain 2 (D2S6). (C) G1451 in the sixth transmembrane helix of domain 3 (DIIIS6), in close proximity to the channel pore. h, Homo sapiens; m, Mus musculus; a, Anolis carolinensis (reptile); f, Takifugu rubripes (fish); dpara, Drosophila melanogaster ‘paralytic’ (alternative name NaV1). Amino acids are indicated by the single-letter code and dots represent identity to the human amino acid. Multiple sequence alignment was carried out using ClustalW V.2.0.12.48
Electrophysiology
Measurements were carried out at the Department of Physiology (Radboud University Medical Center, the Netherlands) with an EPC9 patch clamp amplifier and Patchmaster software (HEKA electronics, Lambrecht, Germany). The sampling interval was set at 50 µs (20 kHz) and low-pass filtering was set to 5.0 kHz in all experiments. Pipettes were pulled from thin-wall borosilicate glass and had resistances between 0.9 and 1.5 MΩ when filled with the pipette solution. Series resistance compensation was set to 85–95% in all experiments. The leak currents were subtracted using a P/6 procedure from a holding potential of −120 mV.
Currents were activated by 40 ms voltage pulses ranging from −70 to +20 mV (in 10 mV increments) from a holding potential of −120 mV at a sweep frequency of 0.2 Hz. Current–voltage curves were converted to conductance–voltage curve, normalised and individually fitted with a Boltzmann equation (see below). Steady-state inactivation was studied by holding cells at conditioning voltage ranging from −120 to −20 mV (in 10 mV increments) for 500 ms and tested with a 40 ms depolarising pulse to −10 mV. The current elicited after the conditioning voltage period was normalised to the current amplitude obtained from a holding voltage of −120 mV obtained before the conditioning period.
The extracellular solution contained (in mM): 132 NaCl, 4.2 KCl, 1 CaCl2, 1 MgCl2, 10 hydroxyethyl piperazineethanesulfonic acid (HEPES), 25 glucose and pH adjusted to 7.4 with NaOH. The pipette solution contained (in mM): 140 CsF, 10 NaCl, 1 ethylene glycol tetraacetic acid, 10 HEPES and pH adjusted to 7.3 using CsOH. After break-in, a 5 min waiting period permitted equilibration between the pipette solution and the cytoplasm of the cell. Current density was calculated by dividing the current (in pA) by the cell capacitance (in pF).
Statistical analysis and fitting of electrophysiological data
Current–voltage curves were converted to conductance and fitted using the following Boltzmann equation: g(V)=gmax/(1+exp((V−V0.5)/k)), where g is the conductance, gmax is the maximal conductance, V is the voltage, V0.5 is the voltage of half-maximal activation and k is the slope factor. Steady-state inactivation curves were fitted with an analogous equation. Data are shown as mean±SEM. The comparisons between mean of the fit parameters and amplitudes were performed using one-way analysis of variance followed by Tukey post hoc test.
Western blotting
To assess protein stability, mutant cDNAs were transfected into HEK293 cells, which lack endogenous sodium channel protein. After culture for 24 h at 37°C, transfected cells were harvested and lysed in radioimmunoprecipitation assay buffer. Aliquots containing 30 μg of protein were incubated with Laemmli sample buffer for 20 min at 37°C, electrophoresed through 4–15% acrylamide gradient gels (Criterion) and immunostained with rabbit polyclonal anti-Nav1.6 (Alomone ASC-009, 1:100). As an internal control, α-tubulin was immunostained with a 1:1000 dilution of monoclonal antibody CLT9002 (Cedarlane Labs, Burlington, Canada).
Results
Clinical phenotypes
In total, 500 patients with ID and 100 patients with a movement disorder were subjected to exome sequencing. Three patients carried mutations of SCN8A. One of them was included in the movement disorders group also, despite having an ID, because of his pronounced movement disorder. The clinical features of these patients are summarised in table 1 and in online supplementary data. Patients 1 and 2 experienced seizures and peripheral nervous system disabilities, but these were not present in patient 3.
Clinical features of three patients with de novo mutations in SCN8A
Mutation detection
In patient 1, a single de novo mutation was revealed after trio-exome sequencing, SCN8A (c.2952C>G; p.(Asn984Lys)). This mutation alters an evolutionarily conserved amino acid adjacent to the transmembrane segment of the voltage-gated sodium channel Nav1.6 (N984K; figure 1). Singleton sequencing of patient 2 and analysis for mutations in a movement disorder gene panel (including genes for ataxia, spastic paraplegia and dystonia) did not identify pathogenic variants. After inspection of the full exome data, an SCN8A mutation was identified as a candidate (c.4351G>A; p.(Gly1451Ser)). The mutated residue is located in a conserved residue of transmembrane segment D3S6 of Nav1.6 (G1451S; figure 1). Sanger sequencing of parental DNA revealed that this mutation had also occurred de novo. In patient 3, two de novo mutations were identified by trio-exome sequencing, SCN8A (c.172G>A; p.(Asp58Asn)) and RING 1 (c.284G>A; p.(Arg95Gln)). This SCN8A mutation alters a residue in the cytoplasmic N-terminus (D58N; figure 1). No likely causative homozygous or compound heterozygous mutations were detected in the exome data of these three patients. Hemizygous mutations were also excluded for patients 2 and 3.
Electrophysiological characterisation of NaV1.6 α subunit mutants
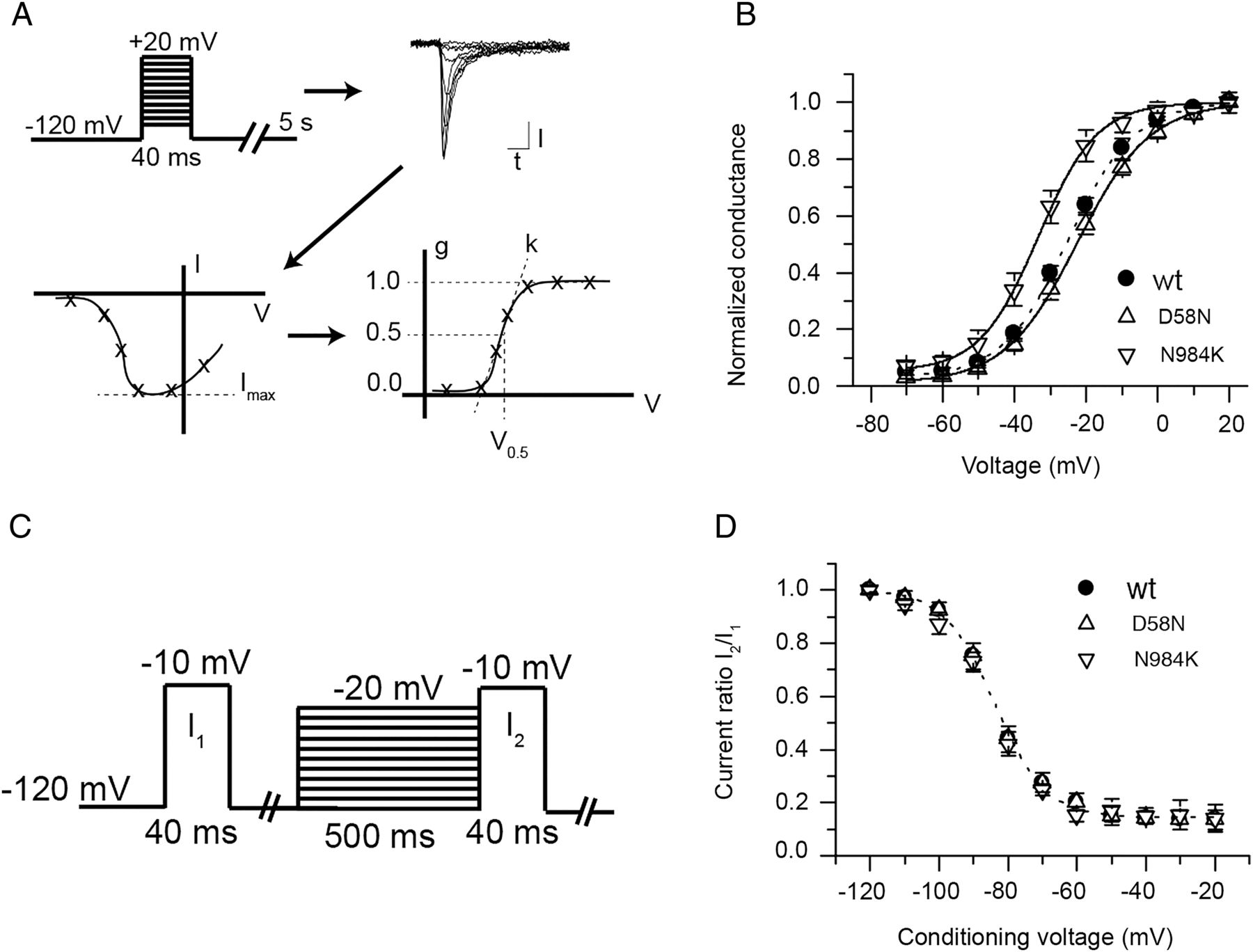
HEK293 cells were transfected with wildtype (wt) or mutated SCN8A cDNA encoding the NaV1.6 α subunit and subjected to whole-cell patch clamp measurements essentially as described previously.36 Cells expressing wt NaV1.6, N984K or D58N demonstrated substantial voltage-evoked currents (figure 2A). Although current amplitudes did not reach their maximal value at the same test voltage, the average maximal inward currents observed for the wt channel and for the D58N and N984K mutants were not statistically different from each other (figure 2B). Current densities in cells expressing the G1451S mutant were ∼10-fold smaller than wt transfected cells and were comparable to non-transfected cells (figure 2A). To determine whether the loss of activity of the G1451S was a consequence of protein instability, as recently reported for the R223G mutation,32 we carried out western blotting. The G1451S mutant proteins were detected in similar levels as wt NaV1.6 in transfected cells (see online supplementary figure S3).

Voltage-evoked currents of mutant proteins in transfected HEK293 cells. (A) Wild type (wt), D58N and N984K gave rise to substantial voltage-evoked currents while the G1451S expressing cells demonstrated current densities similar to non-transfected cells. (B) Average current–voltage curve for non-transfected cells (squares), wt (full circles), D58N (upward triangles), N984K (downward triangles) and G1451S (stars). N=3 for non-transfected cells, and N≥10 for all other conditions.
Wt and D58N mutant channels generated currents with similar voltage of half-maximal activation (V0.5, figure 3B, see 'Methods’ section). Strikingly, the N984K mutant had a 10 mV hyperpolarising shift in V0.5 in comparison with wt (figure 3B). The slope factor k of the Boltzmann fit was not different between conditions (p>0.05, n≥10). Cells were subjected to a steady-state inactivation protocol in order to study fast inactivation (figure 3C and see ‘Methods’ section). Currents from cells expressing wt, N984K and D58N demonstrated no statistically significant differences in voltage dependence of steady-state inactivation or slope (figure 3D).

{kind=link}
{kind=link}
{kind=link}
Voltage dependence of activation and steady-state inactivation of mutant proteins in transfected HEK293 cells. (A) Individual current–voltage (I–V) curves were converted to normalised conductance–voltage (g-V) curves and fitted with a Boltzmann equation yielding the voltage of half-maximal activation (V0.5) and slope factor (k). (B) Average normalised conductances of wild type (wt), D58N and N984K as a function of voltage. Lines represent Boltzmann fit to the average conductance voltage curve. The N984K mutant V0.5 is significantly shifted towards hyperpolarised voltages values (p<0.05, n=10). N≥10 for each conditions. (C) The protocol used to study steady-state inactivation is shown. The fraction of non-inactivated channels after a 500 ms conditioning period is indicated as the ratio of the current after the conditioning period (I2) to the current obtained before conditioning (I1). (D) The average current ratio (I2/I1) for wt, D58N and N984K as a function of the conditioning voltage. The dotted line represents a modified Boltzmann fit to the average wt steady-state inactivation curve. Fit to D58N and N984 is not shown for the purpose of clarity. There is no difference in voltage of half-maximal inactivation between wt and mutants (p>0.05, n≥8).
Discussion
We report three novel de novo missense mutations in SCN8A identified by clinical exome sequencing in patients with ID and variable clinical features. The degree of developmental delay varied from profound ID in patient 1 to moderate ID in patients 2 and 3. Patients 1 and 2 also presented with epilepsy and a movement disorder (including hypertonia, spasticity and ataxia), whereas patient 3 had no such signs. Comparison of the electrophysiological properties of the mutant channels revealed distinct effects.
Patient 1 is most severely affected clinically, with a diagnosis of EIEE that included onset of intractable seizures at 6 weeks of age, feeding by gastric tube and lack of speech. The N984K mutation of SCN8A in this patient resulted in a hyperpolarising shift of ∼10 mV in the voltage dependence of activation (figures 2 and 3), which predicts premature channel opening and hyperactivity of neurons. A similar hyperpolarising shift in voltage dependence of activation of SCN8A was recently reported in a severely affected child with the de novo mutation T767I in transmembrane segment D2S1, seizure onset at 2 weeks of age, profound developmental delay and feeding by gastric tube.28 These two cases, together with the original N1768D mutation,25 demonstrate clearly that gain-of-function mutations of SCN8A that cause elevated channel activity result in severe EIEE.
In patient 2, the G1451S mutation causes apparent loss of channel activity in the HEK cell assay. G1451 is located in a residue of domain 3 that has been suggested to regulate bending of the pore-lining S6 segment during the opening of the channel pore37 and would be expected to alter channel activity. Patient 2 had later seizure onset, at 18 months of age, and severe developmental delay, but could walk and speak at 6 years of age. In a similar case, the partial loss-of-function mutation R223G was described in a patient with seizure onset at 6 months and profound developmental impairment.32 Both of these missense mutations generate full-length protein with reduced channel activity and have a severe clinical phenotype including early-onset seizures. Both patients are more severely affected than the family with an SCN8A protein truncation mutation, which resulted in ID without seizures.24 Two protein truncation mutations have been described in the ESP6500 dataset, but one is an indel of one C nucleotide in a run of four Cs, and the second is a singleton; neither has been confirmed by Sanger sequencing and both must be considered unconfirmed. In three other patients with ID without seizures, loss of SCN8A protein due to contiguous gene deletion has been described; at least two were de novo deletions (http://decipher.sanger.ac.uk). The difference between the protein truncation or deletion patients, and the missense patients, suggests that there may be a dominant effect of the full-length inactive proteins with the G1451S and R223G mutations. The mutant proteins could result in sequestration of interacting proteins such as β subunits, which is required for neurite outgrowth and high-frequency neuronal firing of NaV1.6 expressing neurons.38 Alternatively, there may be gain-of-function features of these proteins that were not detected in the heterologous HEK293 cell assay. Such an observation was made with a mutation of NaV1.1, with apparent loss of function in cultured non-neuronal cells, which was partially rescued in primary cultures of mouse embryo neocortical neurons and exhibited a gain-of-function effect.39 The subcellular localisation of the G1451S mutant of NaV1.6 has not yet been established, and it will be important to examine this mutant in a neuronal cell assay.
The third patient carried the de novo mutation D58N located in the cytoplasmic N-terminal domain of the protein. Electrophysiological analysis revealed full activity of this mutant channel in HEK cells, with no change in biophysical properties. The N-terminal domain of Nav1.6 is likely to function in protein trafficking rather than channel activity. The mutation S21P in the N-terminus of mouse Nav1.6 results in protein retention in the endoplasmic reticulum.40 A binding site for the microtubule-associated protein MAP1B has been mapped between residues 77 and 88 of Nav1.6.41 The activity of the D58N mutant channel in HEK cells demonstrates successful transport to the cell surface, suggesting that this mutation is not pathogenic. However, it is possible that localisation of the D58N channel to the axon initial segment or node of Ranvier in neurons could be impaired. Nevertheless, with the apparent normal function of the D58N mutants in HEK cells, it is also possible that ID in patient 3 might be caused by the second de novo mutation in the RING1 gene encoding a E3 ubiquitin ligase. Mutations in RING1 have not been previously associated with ID. The future detection of similar SCN8A or RING1 mutations may resolve this issue.
Despite the fact that only six SCN8A mutations have been tested functionally, preliminary correlations between mutation type and disease severity are emerging (table 2). First, the de novo SCN8A mutations in patients with EIEE13, the most severe form of SCN8A-related disease, are missense mutations rather than protein truncation mutations.13 Second, three of these missense mutations clearly result in channel hyperactivity, leading to both seizures and ID. Third, complete loss of function due to protein truncation or gene deletion is associated with isolated ID without seizures24 (http://decipher.sanger.ac.uk). In the heterologous test systems used for analysis, two missense mutations associated with ID and epilepsy, G1451S and R223G, exhibit (partial) loss-of-function rather than hyperactivity; these merit additional study to detect possible gain-of-function features.
Functional analysis of SCN8A mutations identified in heterozygous state in patients with intellectual disability (ID) with or without epileptic encephalopathy (EE)
The data for SCN8A are in striking contrast with Dravet syndrome, or EIEE6, which is caused by mutations of the sodium channel SCN1A. In Dravet, 50% of the >800 de novo mutations in sodium channel SCN1A are protein truncation mutations with clear loss of function, and these result in both seizures and ID.42–44 The milder generalised epilepsy with febrile seizures plus phenotype is associated with missense mutations of SCN1A. The difference in the consequences of loss of function may reflect their different roles in inhibitory and excitatory neurons since NaV1.6 (encoded by SCN8A) is the major sodium channel in excitatory neurons while NaV1.1 (encoded by SCN1A) appears to have a critical role in inhibitory neurons.45–47
In summary, we have identified and characterised three novel de novo mutations of SCN8A. Electrophysiological studies indicate that one mutation leads to a gain of function (N984K), the second causes apparent loss of function (G1451S) and the third has normal channel activity and may be non-pathogenic (D58N). These data strengthen previous findings linking gain-of-function mutations of SCN8A with EIEE and demonstrate the importance of functional testing in establishing the pathogenicity of de novo mutations. Functional characterisation of additional SCN8A mutations will provide a more comprehensive understanding of the specific phenotypes associated with gain-of-function and loss-of-function mutations.
Acknowledgments
We thank Janelle O'Brien and Jacy Wagnon for helpful discussions and assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online figures
Footnotes
-
MHM and EJK share senior coauthorship.
-
Contributors MGB has contributed to the acquisition, analysis and interpretation of data, and drafting of the manuscript. MHW, JBW, BPvdW, PP, MCJ and JN contributed to data acquisition, interpretation and revising the manuscript for intellectual content. SD-H, SGW, TK, HGY and RJMB contributed to data interpretation and revising the manuscript for intellectual content. MHM and E-JK are accountable for design of the work, interpretation of data and revision for intellectual content. All coauthors have approved the final version of the manuscript. No other persons contributed in a way that they are entitled to be coauthors.
-
Funding Supported by the National Institutes of Health (R01 NS34509 to MHM). SDH and SGW were supported by grants from the Rehabilitation Research Service, the Veterans Administration, USA.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethics committee of the Radboud University Medical Center.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Except for the raw data, no additional data are available. Raw data may be made available if it does not infringe patients rights.