Article Text

Abstract
First introduced into mammalian organisms in 2013, the RNA-guided genome editing tool CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9) offers several advantages over conventional ones, such as simple-to-design, easy-to-use and multiplexing (capable of editing multiple genes simultaneously). Consequently, it has become a cost-effective and convenient tool for various genome editing purposes including gene therapy studies. In cell lines or animal models, CRISPR-Cas9 can be applied for therapeutic purposes in several ways. It can correct the causal mutations in monogenic disorders and thus rescue the disease phenotypes, which currently represents the most translatable field in CRISPR-Cas9-mediated gene therapy. CRISPR-Cas9 can also engineer pathogen genome such as HIV for therapeutic purposes, or induce protective or therapeutic mutations in host tissues. Moreover, CRISPR-Cas9 has shown potentials in cancer gene therapy such as deactivating oncogenic virus and inducing oncosuppressor expressions. Herein, we review the research on CRISPR-mediated gene therapy, discuss its advantages, limitations and possible solutions, and propose directions for future research, with an emphasis on the opportunities and challenges of CRISPR-Cas9 in cancer gene therapy.
- Clinical genetics
- Gene therapy
- Infection
- Oncology
Statistics from Altmetric.com
Introduction
Gene therapy involves manipulating DNA or RNA for human disease treatment or prevention. The strategies of gene therapy are diverse, such as rectifying, replacing or deleting the culprit genes in genetic diseases, producing disabling mutations in pathogen genomes to combat infectious diseases or inducing therapeutic or protective somatic mutations. It is a promising therapy for a wide range of human diseases including hematological diseases,1 ,2 cancer,3 AIDS,4 diabetes,5 ,6 heart failure,7 and neurodegenerative diseases.8 Up to now, there has been more than 2000 gene therapy clinical trials worldwide,9 with a few gene therapy products having already been approved by authorities, such as Gendicine for head and neck squamous cell carcinoma in China, and Cerepro for malignant brain tumours in Europe.10
Targeted genome editing with programmable nucleases, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), enables diverse genome manipulations in a site-specific manner, such as gene activation/inactivation, sequence deletion, element replacement and chromosomal rearrangement.11 ,12 Unlike previous gene therapy tools that add or insert an exogenous DNA copy into the target cell nucleus or genome, which may give rise to side effects such as insertional mutations and non-physical expression of proteins, the programmable nucleases use a ‘cut-and-paste’ strategy, that is, remove the defect and install the correct, thus representing an preferable tool for gene therapy. Recently, a RNA-guided genome editing tool termed CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9) added to the list of programmable nucleases, offers several advantages over its counterparts and shows therapeutic potentials. Herein, we introduce the basic mechanisms and merits of CRISPR-Cas9 in genome editing, retrospect studies on CRISPR-mediated gene therapy in cell lines and animal models, discuss its challenges and possible solutions and prospect future directions.
Mechanisms and merits of CRISPR-Cas9 in genome editing
First applied in mammalian cells in 2013,13 ,14 CRISPR-Cas9 genome editing tool is adapted from microbial adaptive immune defense system (figure 1A). As shown in figure 1B, the core components of CRISPR-Cas9 are a nuclease Cas9 comprising two catalytic active domains RuvC and HNH, and a single guide RNA (sgRNA) that is derived from CRISPR RNA (crRNA) and trans-acting CRISPR RNA.15 On the presence of a protospacer-adjacent motif (PAM) on the opposite strand, sgRNA directs Cas9 to the target site by base-pairing, resulting in Cas9-generated site-specific DNA double-strand breaks (DSBs) that are subsequently repaired by homologous directed repair (HDR) if the homologous sequences are available or otherwise by non-homologous end-joining (NHEJ).13 ,14 ,15 HDR leads to precise gene correction or replacement whereas NHEJ is error prone and may induce small insert or delete (indel) mutations (figure 1C). Additionally, Cas9 can be reprogrammed into nickase (nCas9) by inactivating either RuvC or HNH,16 or into catalytically inactive Cas9 (termed dead Cas9, or dCas9) by inactivating both of them (figure 1D).17 dCas9 can be repurposed as a site-specific DNA-binding domain transporting various functional domains to the target locus (figure 1D).17
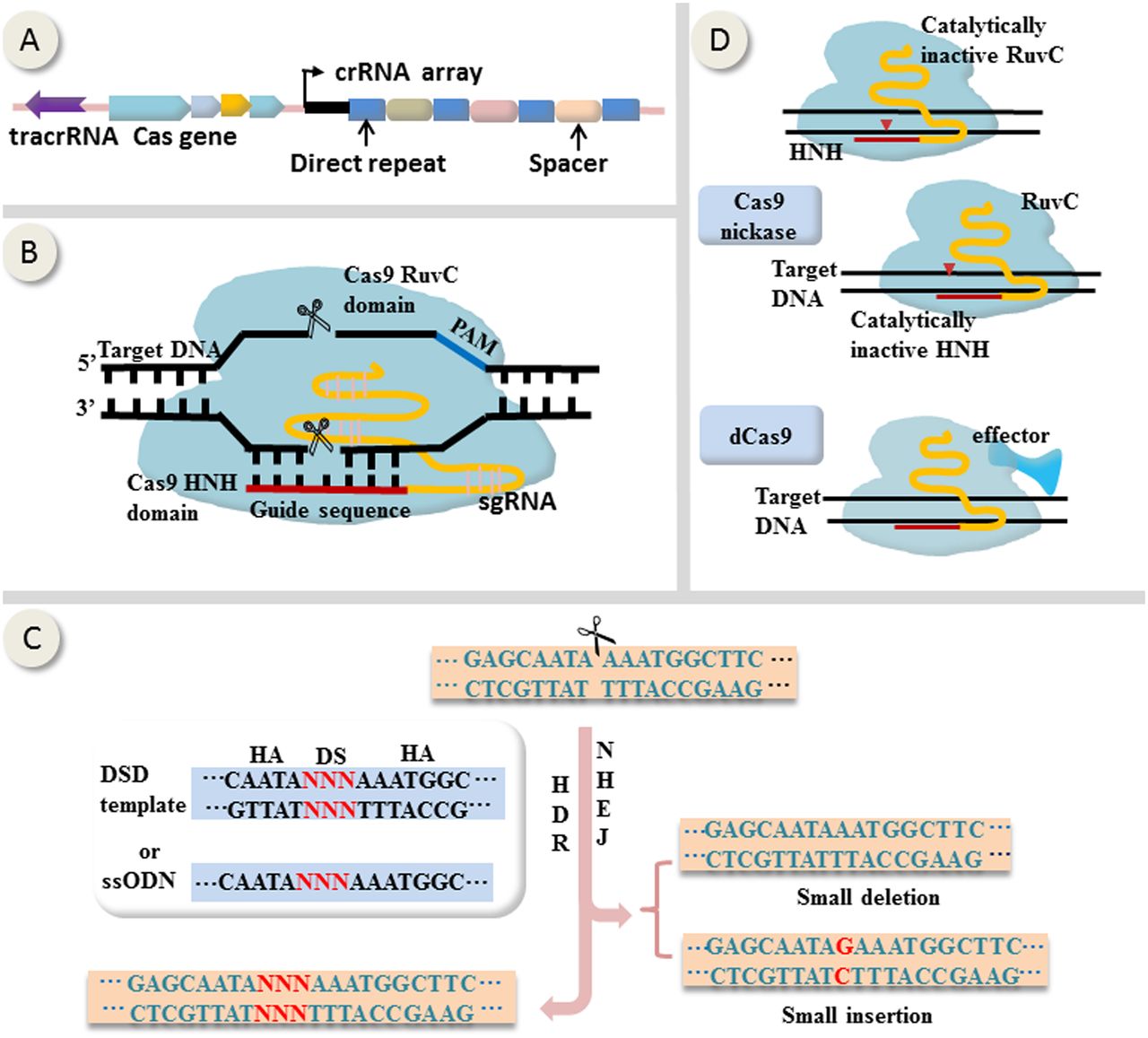
Mechanisms of CRISPR-Cas9-mediated genome editing and epigenome modulation. (A) Representative schematic of CRISPR locus (from Streptococcus pyogenes). (B) Site-specific DNA cleavage by nuclease Cas9 directed by complementary between a single guide RNA (sgRNA) and the target sequence on the presence of a PAM on the opposite strand. sgRNA derives from crRNA and tracrRNA. Wild-type Cas9 possesses two catalytically active domains termed HNH and RuvC, each of which cleaves a DNA strand. (C) Double-strand breaks generated by Cas9 are subsequently repaired by homologous directed repair (HDR) if the homologous sequences are available or otherwise by non-homologous end-joining (NHEJ). Generally, HDR leads to precise gene correction or replacement whereas NHEJ is error prone and may induce small insert or delete (indel) mutations. (D) Cas9 can be reprogrammed into nickase (nCas9) by inactivating either RuvC or HNH, or into catalytically inactive Cas9 (termed dCas9) by inactivating both of them. dCas9 can be repurposed as a site-specific DNA-binding domain fused to various effectors. crRNA, CRISPR RNA; tracrRNA, trans-activating crRNA; HA, homologous arm; DS, donor sequence; DSD, double-strand DNA donor; ssODN, single-stranded oligonucleotide DNA; PAM, protospacer-adjacent motif.
This RNA-guided genome editing tool provides several advantages over conventional protein-guided ones such as ZFN and TALEN. First, with CRISPR-Cas9, to target a new site requires only the design of a complementary sgRNA (the nuclease Cas9 remains the same in all cases), which is much simpler than the de novo synthesis of a bulky guiding protein as in ZFN- or TALEN-based tools. Second, with multiple sgRNAs that target different genomic loci, CRISPR-Cas9 can edit these genomic loci in parallel, a property called multiplexing.13 ,15 However, protein-guided genome editing tools such as TALEN also have some advantages over CRISPR-Cas9. First, the targets of CRISPR-Cas9 are restricted by the presence of PAM sequence and a guanine at the 5′ end 11, whereas the only restriction of TALEN targets is the presence of thymine at the 5′ end,18 which means that TALEN can target more genome sites than CRISPR-Cas9. Second, TALEN are generally reported to produce less off-target effects in relative to CRISPR-Cas9,19 ,20 partially by virtue of the heterodimeric construction of FokI nuclease in TALEN. And moreover, TALEN can be re-engineered to target and cleave mutant mitochondrial DNA in patient-derived cells,21 whereas mitochondrial genome-targeting CRISPR-Cas9 remains an area to be explored. For more details on the comparison among these programmable nucleases, please see review articles such as references 11, 18 and 20.
RNA interference (RNAi) technology is also a convenient and widely used way for the in vivo manipulation of gene expression. However, the effects of RNAi are temporary, non-specific and limited to knocking down transcribed genes. CRISPR-Cas9 system, on the contrary, can induce both gain- and loss-of-function mutations, and suffers from less off-target effects for that CRISPR-Cas9 derives from prokaryote and hence has less crosstalk with eukaryotic components than RNAi pathway does.
With these merits, CRISPR-Cas9 has become as an easy and versatile tool for various genome editing purposes. For example, CRISPR-Cas9 has been used to recapitulate cancer-associated genomic alterations both in vitro and in vivo,22–25 providing a quick and convenient platform for functional investigations of cancer-related genetic mutations. Recently, a more exiting area, that is, the application potentials of CRISPR-Cas9 in gene therapy, has gradually emerged.
CRISPR-Cas9-mediated gene therapy in cell lines and animal models
Engineering pathogen DNA to combat infectious diseases
CRISPR-Cas9 is derived from microbial adaptive immune defense system, and thus possesses inherent advantages in engineering and disabling pathogen genomes to treat infectious diseases.
In August 2013, just 7 months after the CRISPR-Cas9 genome editing tool was first introduced into mammalian cells,13 ,14 Ebina et al26 reported that CRISPR-Cas9 can mutate long terminal repeat (LTR) sequence of HIV-1 in vitro, resulting in removal of the integrated proviral DNA from the part of the host cells and a significant drop in virus expression. To our knowledge, this study is the first one to experimentally explore the gene therapy potentials of CRISPR-Cas9 system. Recently, another independent study harnessed CRISPR-Cas9 to mutate HIV-1 LTR U3 region and gained similar results.27
Chronic hepatitis B is one the most common infectious diseases worldwide, which can lead to liver cirrhosis and cancer. The persistence of hepatitis B virus (HBV) infection is due to the intrahepatic existence of HBV covalently closed circular DNAs (cccDNAs), which are hard to clear by present therapeutics and serve as a reservoir for HBV reactivation. Recently, a study by Seeger and Sohn28 showed that in HepG2 cells expressing HBV, the introduction of cccDNA-targeting CRISPR-Cas9 system resulted in both decreased hepatitis B core antigen expression and mutated cccDNAs, providing impetus for further research on the possibility of CRISPR-Cas9-mediated cccDNA clearance.
Correcting monogenic disorders
Monogenetic disorder is caused by single gene defects. Compared with polygenic diseases such as cancer, monogenic disorders are more amenable to gene therapies. Currently, the correction of monogenic disorders represents the most translatable field in CRISPR-Cas9-mediated gene therapy.
Gene correction in animal germline
Germline modification is a conventional approach for gene therapy studies in animal models, though it is currently not applicable to humans.
In a mouse model of Duchenne muscular dystrophy (DMD), an inherited X-linked monogenic disease, CRISPR-Cas9-mediated gene editing in the germline gave rise to genetically mosaic offsprings with 2–100% somatic cells carrying the corrected version of the culprit gene.29 Intriguingly, the extent of phenotype rescue surpassed the percentage of gene correction, for example, 17% gene corrections resulted in 47–60% rescued muscle cells in one of the experimental mouse, indicating a growth advantage imposed by the gene correction.
Study by Wu et al30 serves as another example. One basepair deletion in exon 3 of Crygc (crystallin gamma C) gene in mouse causes frameshift mutation and cataract phenotype. In such a mouse model, germline-manipulation with CRISPR-Cas9 system was capable of correcting both the mutant gene and cataract phenotype in part of offsprings. Briefly, plasmids expressing Cas9 mRNA and a sgRNA (targeting the defect Crygc gene) were injected into murine zygotes carrying Crygc mutation. Of the 22 live offsprings, 10 underwent culprit gene modifications (the editing efficiency is 45.45%) either by HDR (n=4) (figure 1C left) or by NHEJ (n=6) (figure 1C right). As expected, all of the four offsprings undergoing HDR were free of cataract due to HDR-mediated gene corrections. Interestingly, however, of the six offsprings undergoing NHEJ, two were also free of cataract thanks to coincidental frameshift restoration resulting from NHEJ-induced indel mutations, while the rest four remained cataract phenotype.30 These results highlighted the importance of HDR in reliable gene corrections. However, as demonstrated in this study, zygote manipulation can only rescue part of the offsprings in an unpredictable manner. To gain 100% rescue efficiency, Wu et al31 corrected the culprit gene Crygc in mouse spermatogonial stem cells (SSCs) with CRISPR-Cas9, and then picked out SSC lines that carried the corrected gene without undesired genome perturbations. The pups generated by these preselected SSCs were unexceptionally cataract free.
Somatic gene correction in adult animals
CRISPR-Cas9 technology can also mediate somatic gene corrections in adult animals, bypassing embryo manipulations. Yin et al,32 for example, delivered CRISPR-Cas9 agents and a homologous donor template (in order to increase HDR rate) into adult mice with hereditary tyrosinemia via tail-vein hydrodynamic injection, resulting in gene corrections in 0.25% of liver cells initially, and in 33.5% of liver cells 33 days postinjection (possibly due to selective advantages imposed by the correction), which was sufficient to rescue the disease phenotype. This method is more translatable to human therapeutics because it does not involve embryo manipulations.
Gene correction in human stem cells or induced pluripotent stem cells
Besides in animal models, CRISPR-Cas9 can also exert therapeutic functions in human stem cells or induced pluripotent stem cells (iPSCs). In human intestinal stem cells collected from patients with cystic fibrosis, the culprit gene cystic fibrosis transmembrane conductance regulator was rectified by homologous recombination during CRISPR-Cas9 genome editing while the pluripotency was retained as demonstrated by formations of organ-like expansions in cell culture.33 The clinical translatability of this study gets support from previous study34 in which ex vivo cultured colon organoids were successfully transplanted into mice colons and formed functionally and histologically normal crypts.
Non-multipotent somatic cells can be reprogrammed into iPSCs, with multipotentiality and self-renewal ability similar to embryonic stem cells. iPSC represents an ideal cell tool for gene therapy because patient-derived iPSC can be genetically modified in vitro, then differentiates into desired cells for therapeutic autologous transplantation. With CRISPR-Cas9, gene corrections have been successfully conducted in iPSCs derived from β-thalassemia patients35 and DMD patients,36 paving the way for ultimate clinical applications of gene therapies for these two inherited diseases based on CRISPR-Cas9 and iPSC.
The above-mentioned monogenic disorders are all caused by loss-of-functions mutations. In monogenic disorders that are resulted from gene duplications (eg, Friedreich's ataxia), or in autosomal dominant disorders in which the affected genes are haplosufficient, CRISPR-Cas9 also holds therapeutic potential given its ability in gene deletion23 and inactivation.24 ,25
Inducing therapeutic or protective mutations
In the treatment of non-genetic or polyetiological diseases, CRISPR-Cas9 is also a potential participant by inducing therapeutic or protective mutations in somatic tissues. For example, mutation in CCR5 (CC chemokine receptor 5) gene that leads to cell-resistance to HIV-1 infection was achieved in iPSCs with CRISPR-Cas9.37 Interestingly, a recent clinical trial38 showed that autologous reintroduction of CD4T cells whose CCR5 gene was inactivated in vitro by ZFN was safe and lead to decreased viral load, lending support to the potential of CRISPR-Cas9 in AIDS gene therapy.
Another example involves the PCSK9 (proprotein convertase subtilisin/kexin type 9) gene, whose spontaneous loss-of-function mutations in a small fraction of human beings are associated with lower plasma low-density lipoprotein (LDL) level39 In mice liver, CRISPR-Cas9 was reported to successfully induce loss-of-function mutation at PCSK9 gene locus, leading to elevated hepatic LDL receptor level and dropped plasma cholesterol level, thus holding therapeutic potentials for hypercholesterolaemia, a common disorder in modern society.40
CRISPR-mediated cancer gene therapy
Although only very few gene therapy products were licensed for cancer treatment during the past decades,3 gene therapy remains one of the hottest spots in cancer research nowadays, given that cancer is a genetic disease and that gene therapy holds the promise of combating cancer from inside with minimum side effects. Of the more than 2000 gene therapy clinical trials worldwide, 64.1% were cancer gene therapy.9
Inactivation or clearance of oncogenic virus
Many viral infections are associated with carcinogenesis, such as HBV and hepatitis C virus in liver cancer, Epstein-Barr virus (EBV) in nasopharyngeal carcinoma and human papillomavirus (HPV) in cervical cancer. Clearance or inactivation of these oncogenic viruses can interrupt and may even reverse tumorigenesis process. CRISPR-Cas9 editing system is derived from bacterial native immune system and hence has inherent advantage in defense against or clearance of viral infection. Examples have already existed of CRISPR-Cas9-mediated antivirus and antiproliferation effects in HPV-positive cervical carcinoma cell line41 and EBV-positive Burkitt's lymphoma cell line42 Zhen et al41 reported that the transfection of Cas9 and sgRNAs targeting HPV16 E6 or E7 into HPV-16-positive cervical carcinoma cell lines SiHa resulted in the inhibition of cancer cell proliferation both in vitro and in xenograft mouse model. Similarly, in a Burkitt's lymphoma cell line with latent EBV infection, CRISPR-Cas9 treatment inhibited cell proliferation and reduced viral load.42 Therefore, CRISPR-Cas9-mediated inactivation or clearance of oncogenic virus provides a promising and cost-effective option for the prevention and treatment of virus-associated cancers.
Manipulating cancer genome or epigenome for therapeutic purposes
Given that cancer is a genetic disease and that CRISPR-Cas9 is a versatile genomic editing tool, it is easy for us to conceive that to correct oncogenic genome aberrations via CRISPR-Cas9 may represent a promising anticancer strategy. CRISPR-Cas9 can correct genetic mutations as demonstrated in monogenic diseases,30–36 and modulate epigenetic states.16 ,43–46 As two sides of one coin, genetic mutations and epigenetic alterations interact with each other and cooperate to drive cancer initiation and progression. Catalytically inactivated dCas9, for example, can bind to DNA elements and suppress their transcriptional activities, a process called CRISPRi.43 ,44 Moreover, dCas9 can be harnessed as a site-specific DNA binding domain and fused to epigenetic modifiers. Under the guidance of sgRNAs, dCas9 and the modifiers get access to the target sites and exert epigenetic regulations,16 ,45 ,46 a process termed epigenome editing, which is also a potential tool for anticancer modalities. This dCas9-effector strategy has already been applied in genome wide screening study in which gain-of-function mutations in melanoma cells that confer drug resistance were identified.46
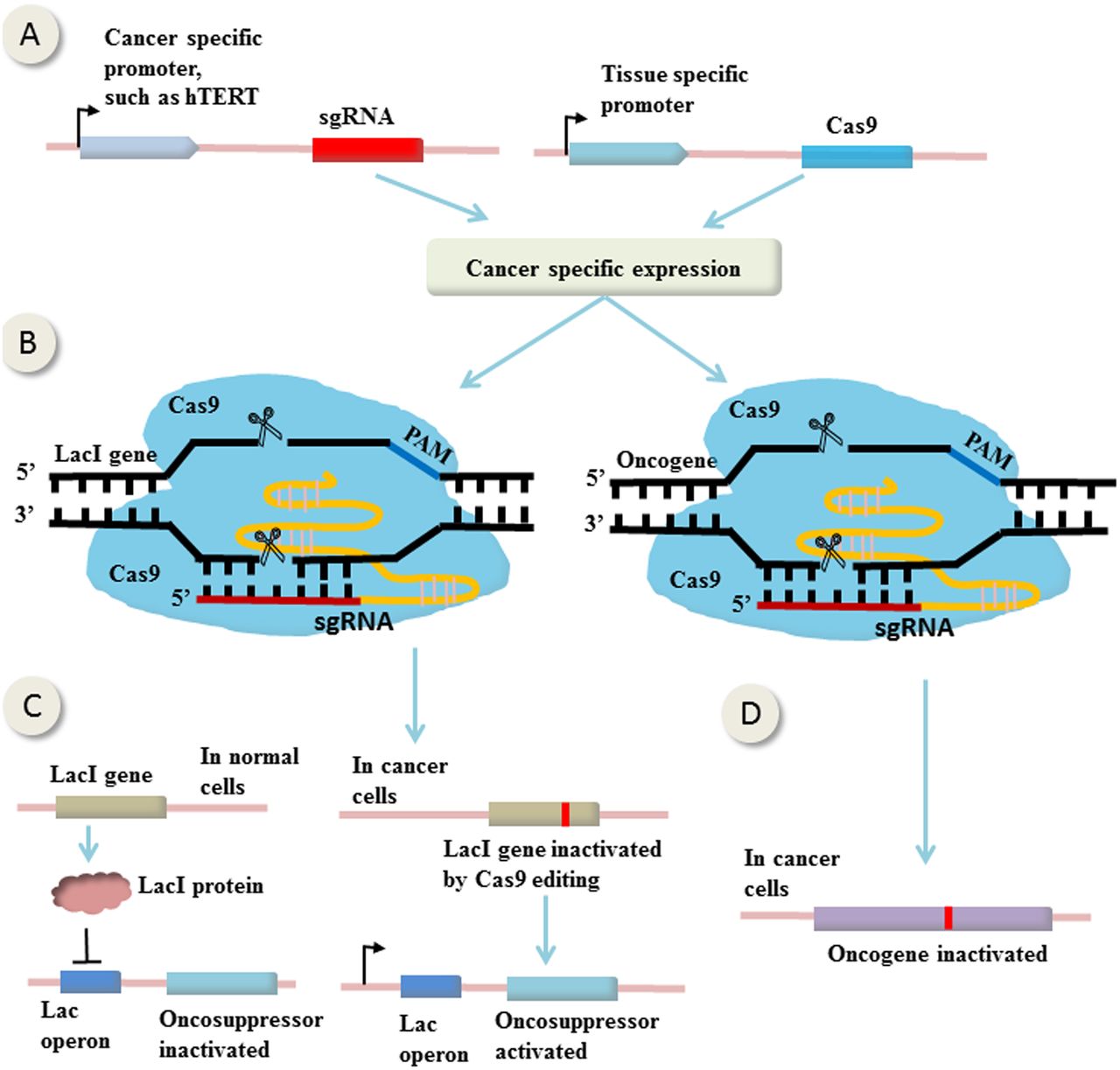
An example of therapeutic genome manipulations comes from study by Liu et al47 in which CRISPR-Cas9-mediated bladder cancer cell-specific genome editing using an AND logic gate (figure 2). It is called ‘AND’ because there are two inputs and a single output, and only the coexistence of the two inputs can generate an output. In this study, the expression of sgRNAs and Cas9 were controlled by two promoters (two inputs), the cancer-specific human telomerase reverse transcriptase promoter and urothelium-specific human uroplakin II promoter (figure 2A), respectively. The fact that only in bladder cancer cells exist both the two promoters enables bladder cancer cell-specific expression of both Cas9 and sgRNAs (figure 2B, left). The sgRNAs were designed to target LacI gene, which expresses LacI protein that binds to Lac operator and hence suppresses the expression of the downstream effector gene (the output) (figure 2C, left). In bladder cancer cells, Cas9 and sgRNAs were both expressed, mutating LacI gene and abrogating its suppression on the effector gene, leading to bladder cancer cell-specific expression of the effector (figure 2C, right). In this study, the effector genes were p21, E-cadherin and Bax (BCL2-associated X), leading to cancer-specific growth inhibition, migration suppression and apoptosis, respectively. Theoretically, the effector can be any oncosuppressors (figure 2C) or oncogenes (figure 2D), or suicide genes that cause tumour inhibition or destruction.

{kind=link}
{kind=link}
CRISPR-Cas9-mediated gene (in)activation in cancer cells. (A) The expression of single guide RNAs (sgRNAs) and Cas9 are controlled by cancer-specific promoter and tissue-specific promoter, respectively. Consequently, only in the specific cancer cells can express both of them. (B) The sgRNAs are designed to target LacI gene or oncogene. (C) In normal cells, LacI protein expressed by LacI gene binds to Lac operator and suppresses the expression of the downstream effector gene. In specific cancer cells, Cas9 and sgRNAs are both expressed, mutating LacI gene and abrogating its suppression on the effector gene, leading to its cancer-specific expression. (D) Loss-of-function mutation of oncogene in cancer cells induced by CRISPR-Cas9 editing. (This figure is partially based on the study by Liu et al47).
However, CRISPR-Cas9-mediated (epi)genetic modulations for cancer therapy suffer from several limitations. First, cancer is a polygenic and heterogeneous disease. Genomic aberration profiles are different in tumours among patients and in tumours during different stages or from different sites within a patient, which renders (epi)genome manipulation therapy in cancer a highly tailored and dynamic process relying on spatiotemporal contents. In current conditions, it is a mission hard to accomplish, even with the assistance of CRISPR-Cas9 system. Second, unlike in monogenic diseases that can be rescued at an initial gene correction efficiency as low as 0.25%,32 effective (epi)genome manipulation therapy in cancer requires fairly high edit efficiency because the unedited cells retain unreduced malignancy, possess selective advantages over the ‘corrected’ cells and proliferate quickly, rendering gene therapy ineffective shortly. However, such a high edit efficiency is still beyond the reach of current CRISPR-Cas9 technology.30 ,32
Challenges and possible solutions
Off-target effects
Because CRISPR-Cas9 causes permanent genome alterations, its off-target effects must be accurately profiled and controlled when applied in gene therapy. Current off-target identification methods comprises mainly of silicon prediction and in vitro selection, which are based on the complementarity between sgRNAs and potential off-target sequences. However, silicon methods can only identify part of the off-target cleavage.48 And moreover, DNA binding and cleavage by Cas9 are in some cases uncoupled, that is, Cas9 can bind to but not cleave DNA sequences that are partially complementary to sgRNA49 and exert epigenomic regulatory effects,43 ,44 which is unpredictable by silicon methods relying on base pairing. So, more unbiased methods are needed to provide a comprehensive off-target profile, such as genome-wide characterisation of Cas9 binding profile by chromatin immunoprecipitation sequencing analysis,49 and genome-wide identification of Cas9 cleavage profile by GUIDE-seq.48
Several methods have been developed to reduce the off-target effects. First, both the structure and composition of the guide RNA can affect the frequency of off-target effects.50 To select a target site that has no homologous sequence throughout the genome is a practical way for reducing off-target effects. Second, the using of a pair of Cas9 nickases (a mutant form of Cas9 that generates single-stranded break rather than DSB) to generate paired nicks on the two strands of target sequence can significantly increase target specificity because off-target single nicks are faithfully repaired.51 It is estimated that this double nicks strategy can increase target-site specificity by about 1500 times.15 Third, sgRNA truncated by 2–3 nt are reported to reduce the off-target effects possibly because shorter sgRNA sequence has a decreased mismatch tolerance.52 Fourth, delivery vehicles by which Cas9 and sgRNAs enter into cells can also affect on- to off-target activity ratio. Cell-penetrate-peptide-mediated delivery was reported to achieve higher on- to off-target activity ratio compared with plasmid-mediated delivery.53 And last, the combination of CRISPR-Cas9 with other nuclease may also help. Tsai et al54 reported that fusing FokI nuclease to dCas9 generates a dimeric RNA-guided FokI nuclease that has an improved specificity compared with wild-type CRISPR-Cas system.
Editing efficiency
Delivery and editing efficiency has long been a crux of gene therapy applications,4 ,55 ,56 especially for cancer. As stated above, cancer gene therapy necessitates high editing efficiency, which is still beyond the reach of current CRISPR-Cas9 technologies. The solutions of this problem lie in future the development of more efficient delivery vectors, more powerful sgRNA, and more potent Cas9. Adeno-associated virus (AAV) vectors are commonly used gene delivery tools for gene therapy research and clinical trials because of their efficiency and safety.1 ,57 ,58 However, the commonly used Cas9 gene derived from Streptococcus pyogenes is too big to transduce with wild-type AAV due to its limited genetic cargo. Using smaller Cas9 orthologs59 derived from other microbes or engineering AAV to increase its genetic cargo57 may represent feasible ways to enhance delivery efficiency. Though cell-penetrating peptide-mediated delivery of CRISPR-Cas9 into cells was reported to increase editing efficiency, the optimal editing efficiency was still under 20% in vitro.59 The delivery of purified recombinant Cas9 protein instead of Cas9 gene into human cell line, on the other hand, can achieve an editing efficiency as high as 79%.60
HDR rate
The DSBs produced by CRISPR-Cas9 are subsequently processed by NHEJ or HDR. In many cases, HDR results in desired gene modifications while NHEJ gives rise to undesired indels that cause uncontrollable gene disruptions. As shown in the study by Wu et al30 among mice genetically edited by CRISPR-Cas9, all HDR-mediated editing corrected the causative gene defects and rescued disease phenotype while only a small part of NHEJ-mediated editing had the corrective effect. Therefore, increasing HDR rate can improve the efficiency and reliability of CRISPR-Cas9-mediated gene therapy while decrease genomic toxic effects concurrently.
Cell cycle phase dominates the choice between NHEJ and HDR, with HDR taking place mainly in S and G2 phases due to the availability of sister chromatids as templates.61 Nature of the donor DNA also influences HDR rate.61 ,62 Via cell cycle synchronisation techniques, Lin and colleagues63 conducted timed delivery of Cas9 protein and sgRNA complexes into human cells and gained HDR rate up to 38%. The authors also demonstrated that longer homologous arm and single-stranded oligonucleotide DNA template (compared with double-strand DNA templates) can increase HDR rate. Delivery vector is another influential factor of HDR rate. In a study by Holkers et al,64 CRISPR-Cas9 and DNA donor delivered via protein-capped adenoviral vector resulted in higher HDR rate versus via integrase-defective lentiviral vector or non-viral vector templates. Moreover, previous study showed that bacterium Deinococcus radiodurans can repair its genome form hundreds of short DNA fragments resulting from exogenous damages. This highly efficient DNA repair systems in microbes might be explored and harnessed in the future to increase HDR rate in eukaryote systems.65
Perspectives and conclusion
Compared with monogenetic diseases, studies on CRISPR-mediated gene therapy in polygenic and polyetiologic diseases lag behind. To prospect future applications of CRISPR-Cas9 technology in gene therapy for all human diseases is infeasible in a single review. Herein, we select cancer as an example to discuss future directions.
As stated above, CRISPR-Cas9 based (epi)genome manipulation therapies in cancer are hampered by heterogeneity in oncogenic mutation profiles and the requirement of pretty high editing efficiency. Inactivation or clearance of oncogenic virus with CRISPR-Cas9 is a feasible option but this strategy is limited to virus-associated cancer types and has limited therapeutic efficacy because on initiation, cancer can progress, at least partially, independently of the underlying viral infection. On the other hand, oncolytic virotherapy or host genome modification may represent more promising modalities.
Some virus can be genetically engineered to specifically infect and replicate in cancer cells, killing them through virus-mediated cytotoxicity or enhanced anticancer immune response. Oncolytic virotherapy is one of the most promising fields in cancer gene therapy. Many oncolytic viruses have been tested in clinical trials,66 with one of them licensed in China.67 CRISPR-Cas9 can play a part in oncolytic virotherapy in several ways, such as adding a cancer-specific promoter to genes that are indispensable for viral replication, or inducing mutations in viral genome whose defects can be complemented by cancer-specific metabolites.
Currently, another hot spot in cancer gene therapy is the modifications of the host cells to enhance anticancer immune responses,68 or to increase resistance to chemo- and radiotoxicity.3 Adoptive T-cell therapy, for example, in which genetically engineered T cells were re-infused back into patients, has shown survival improving effects in clinical trials.69 Given that CRISPR-Cas9 has been applied in T cell modification70 and immune modulation,70 ,71 it is reasonable for us to envision its application in cancer immune therapies.
In conclusion, the RNA-guided genome editing tool CRISPR-Cas9 offers several advantages over protein guided counterparts and RNAi techniques. It has shown therapeutic potentials in cell lines or animal models for infectious diseases, monogenic diseases and cancer. Before it is translatable to the clinic to benefit patients, several challenges must be overcome, such as ways to reliably profile and control the off-target effects, ways to increase editing efficiency and ways to improve HDR rate. However, with the rapid advance in CRISPR-Cas9 technology, we can still optimistically anticipate that, in the future, it may revolutionise gene therapy research and become a convenient and versatile tool for human gene therapy practice.
References
Footnotes
-
Lu Xiao-Jie and Xue Hui-Ying are co-first authors.
-
Correction notice This article has been corrected since it published Online First. The fifth sentence in the Editing efficiency section has been corrected to read ‘Using smaller Cas9 orthologs...’
-
Contributors CJ-L and JL-J conceived the idea and collected literatures; LX-J and KZ-P read through and analysed literatures; LX-J wrote the manuscript while XH-Y and JL-J revised it.
-
Funding National High Technology Research (863) Project of China (2012AA020204).
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.