Article Text

Abstract
Background Inactivating germline mutations in the tumour suppressor gene BRCA1 are associated with a significantly increased risk of developing breast and ovarian cancer. A large number (>1500) of unique BRCA1 variants have been identified in the population and can be classified as pathogenic, non-pathogenic or as variants of unknown significance (VUS). Many VUS are rare missense variants leading to single amino acid changes. Their impact on protein function cannot be directly inferred from sequence information, precluding assessment of their pathogenicity. Thus, functional assays are critical to assess the impact of these VUS on protein activity. BRCA1 is a multifunctional protein and different assays have been used to assess the impact of variants on different biochemical activities and biological processes.
Methods and results To facilitate VUS analysis, we have developed a visualisation resource that compiles and displays functional data on all documented BRCA1 missense variants. BRCA1 Circos is a web-based visualisation tool based on the freely available Circos software package. The BRCA1 Circos web tool (http://research.nhgri.nih.gov/bic/circos/) aggregates data from all published BRCA1 missense variants for functional studies, harmonises their results and presents various functionalities to search and interpret individual-level functional information for each BRCA1 missense variant.
Conclusions This research visualisation tool will serve as a quick one-stop publically available reference for all the BRCA1 missense variants that have been functionally assessed. It will facilitate meta-analysis of functional data and improve assessment of pathogenicity of VUS.
- Cancer: breast
- Clinical genetics
- Molecular genetics
- BRCA1
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Breast cancer is the most common cancer in women and approximately 5%–10% of breast cancer cases are hereditary, where a germline mutation is present within a known cancer susceptibility gene.1 Individuals with inherited inactivating mutations in BRCA1 have an increased risk of developing breast and ovarian cancers.2–4 A recent prospective study of mutation carriers estimates the cumulative risk of breast cancer to be 60% (95% CI 44% to 75%) and of ovarian cancer to be 59% (95% CI 43% to 76%) by the age of 70 years.5 Inherited mutation in BRCA1 accounts for 40%–45% of all hereditary breast cancer cases but for around 80% of cases in families with multiple cases of breast and ovarian cancers.6 Thus, women with a strong family history are referred to genetic testing.
BRCA1 genetic test is based on direct sequencing of coding exons7 and can have the following outcomes: no sequence alteration, pathogenic variant, non-pathogenic variant or variants of unknown significance (VUS).8 Despite early evidence to the contrary, it seems that individuals from BRCA mutation-positive families with no sequence alteration or with non-pathogenic variants are not at increased risk for breast and ovarian cancer.9–12 Individuals testing positive for a pathogenic variant have a well-established elevated risk of developing breast and ovarian cancers.5 However, for those that carry a VUS, test results cannot be used for risk assessment. Most of these VUS are alterations in intronic and regulatory regions or in-frame deletions/insertions and missense variants whose impact on function cannot be inferred from the sequence alone.13
Currently, a multifactorial statistical model classifies BRCA variants where each variant is assigned a prior probability of being pathogenic.14–17 This model uses data from segregation analysis, co-occurrence, family history and tumour pathology and can be expanded to take into consideration other sources of data. To align variant classification to specific medical recommendations, the International Agency for Research on Cancer (IARC) Unclassified Genetic Variants Working Group defined a five-class system with specific probability thresholds.18 Systematic use of this model has led to the classification of a large number of previously unclassified variants.16 ,19
However, despite a wealth of information from functional assays, these data have not yet been incorporated into the multifactorial model. Recently, a computational approach to infer disease relevance from functional data, called VarCall, was developed.20 This latest development allows the systematic integration of functional data into the multifactorial model. To facilitate this analysis, we aggregated all data on functional assays for BRCA1 missense variants into one easy-to-use lookup tool to effectively determine the functional significance of the documented VUS.
We call this web tool ‘BRCA1 Circos’ and it currently represents ∼700 documented missense variants (Breast Cancer Information Core (BIC), http://research.nhgri.nih.gov/bic/). Circos plots are visualisation tools widely used in the current literature to represent multidimensional genomic data21 that were adapted by us to display the BRCA1 missense variants. Besides the visualisation resource, we have developed a single comprehensive database that incorporates all the present information on BRCA1 missense variants. We used PERL-based Circos modules to generate the BRCA1 Circos images and developed a webpage for the Circos to be used as an interactive tool along with a searchable database of all the variants (the webpage can be viewed at http://research.nhgri.nih.gov/bic/circos/).
Methods and Results
Literature curation
We annotated all the missense variants that had been reported in (a) the literature or (b) the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP6500SI-V2 release; http://evs.gs.washington.edu/EVS/) or (c) the BIC database totalling 698 variants. We performed an extensive literature search for all functional assays. If the variants were present in the BIC, we have indicated the frequency of the variant in Circos. In addition to the traditional search tools and databases such as the National Center for Biotechnology Information's PubMed and Google Scholars, we also used publically available databases such as Align-GVGD (http://brca.iarc.fr/PRIORS/BRCA1/index.php) and the BRCA1 variant literature database (http://chromium.liacs.nl/LOVD2/cancer/home.php) to document the information available on missense variants in these databases. The information from these sources was used to develop a database of these variants and, when available, the impact on BRCA1 function as indicated in the original source. Online supplementary table S1 contains the complete BRCA1 Circos database. Updated versions will be available for download on the BRCA1 Circos webpage.
Harmonising scores
Regardless of whether the data we obtained from the articles defined the results categorically or quantitatively, we have followed a controlled vocabulary16 to define functional impact or no functional impact as mentioned by the authors in the respective papers. As a rule, we respected the authors’ classification/score of individual variants. For example, in a quantitative test, we followed the authors’ cut-off to determine which variant had a functional impact and which did not. In some cases, we might disagree with the final classification by the authors; however, without access to the raw data for each individual article to allow for reanalysis and without the benefit of the authors’ expertise in the specific assays, we have decided to consistently follow the authors’ determination.
To harmonise scores across all articles, we use controlled vocabulary previously described.8 ,16 We classified variants as having a ‘functional impact’ or ‘no functional impact’ and intermediate phenotypes with ‘mild functional impact’ or ‘intermediate functional impact’. Although many papers report a categorical scoring system (eg, impact vs no impact), others report a continuous scoring system (eg, as percentage of wild-type activity) with a defined cut-off value. However, most articles with continuous data did not have readily accessible raw data, but rather figures with histograms that are not amenable to data extraction and reanalysis. Thus, we decided to use a categorical (and not quantitative) scoring system for consistency. Functional impact has been annotated as follows: N, no functional impact; F, functional impact; I, intermediate functional impact; M, mild functional impact and U, undetermined functional impact on the BRCA1 protein. The ‘intermediate’ and ‘mild functional impact categories’ were created to accommodate cases where intermediate values were reported. For example, in the study by Morris et al,22 BRCA1 Ubiquitin ligase activity, binding to E2 and BARD1 binding was tested. The phenotype for each of these functional studies was assigned as (+++), (++), (+), (−) or (−/+). In this case, (+++) was defined as ‘no functional impact’, (++) as ‘mild functional impact’, (+) as ‘intermediate functional impact’, (−) as ‘functional impact’ and (−/+) as undetermined. As described above, we were always guided by the authors’ interpretation.
It is important to note that functional assays or predictions from protein sequences and structures can only assess a variant's impact on the protein function and not on pathogenicity, a clinical phenotype. Although there is a strong correlation between defects in protein function and pathogenicity, it is conceivable that not all variants that abrogate a certain function will lead to increased risk. The particular function being assayed may be unrelated to the protein tumour suppressor function. Conversely, a variant with no detectable impact on a specific function may have dramatic effect on a critical function of BRCA1 that is not the one being assessed.
Circos plot
Circos (http://circos.ca) is a freely available web resource primarily used to represent genomic data.21 Circos provides PERL-based modules that can be used as the basis to develop circular visualisation of data. We used these modules to install Circos and wrote our own script to represent the data present in our BRCA1 missense variant database. In Circos, each concentric ring is called a ‘track’.
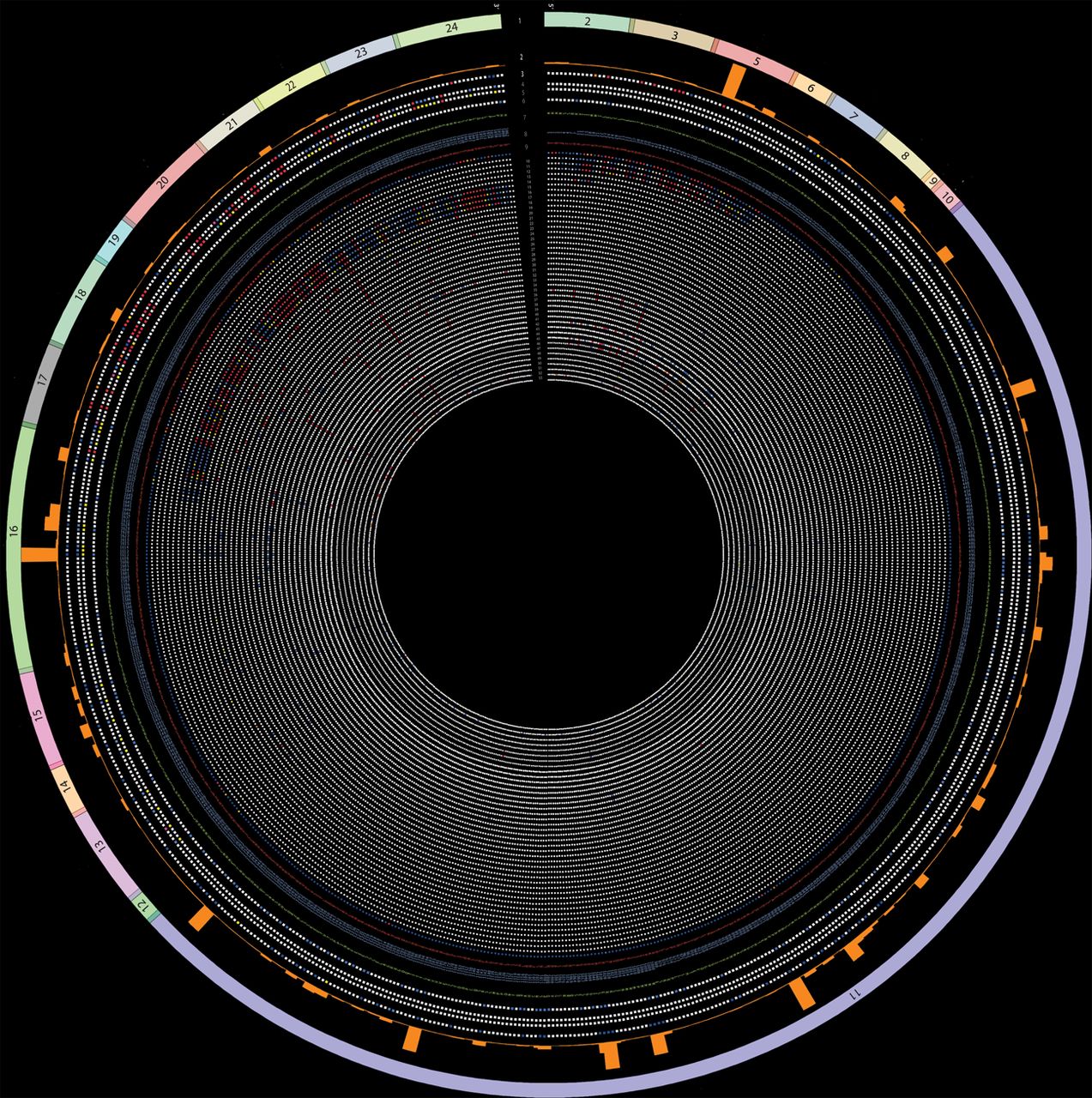
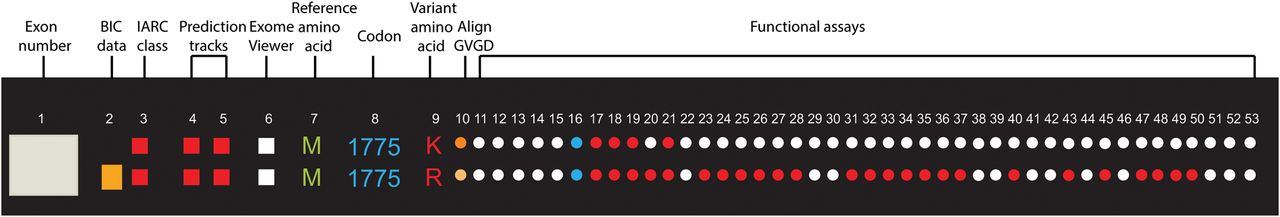
Figure 1 shows all the exons of the BRCA1 gene in the outermost track (track 1). Tracks 2 and 3 represent the frequency of the variant in the BIC database and the IARC class, respectively. Tracks 4 and 5 depict results from the VarCall computational model, with track 4 representing the likelihood of being pathogenic and track 5 showing the associated Bayes’ factor. Track 6 indicates whether the variant is present in the NHLBI Exome Sequencing Project.23 A brief description for each track can be seen in table 1 and detailed descriptions and the key to interpreting them can be found in the online supplementary material 1 (http://research.nhgri.nih.gov/bic/circos/trackdescriptions/). Missense variants are shown in tracks 7–9 (reference amino acid: track 7, codon: track 8, variant amino acid: track 9). The remaining tracks show the functional impact on the variant as assessed by different functional assays (tracks 11–53). The functional assessment by each assay/prediction algorithm is colour coded as ‘red’ (functional impact), ‘blue’ (no functional impact), ‘yellow’ (mild/intermediate functional impact) or ‘white’ (no data available).

BRCA1 Circos excluding exon 11. (A) BRCA1 Circos with no data on display for exon 11 to facilitate data visualisation for the remaining exons (see figure 2 for BRCA1 Circos including exon 11 data). All documented BRCA1 missense variants are represented in the Circos plot that contains 53 concentric tracks (left panel). (B) Magnified view of the region depicted in the inset (dashed lines) from (A). Refer to table 1 for detailed descriptions of each track. BRCA1 Circos can be accessed via the website http://research.nhgri.nih.gov/bic/circos/ with mouse-over and search functionalities.
Track description for BRCA1 Circos
The script generates an image file that was used as the basis for the webpage. We generated two Circos figures: one with all variants in all coding exons (figure 2) and one removing data from exon 11 of BRCA1 (figure 1). This was done because a large number of missense variants in exon 11 have been identified, but the functional data on these variants are sparse. Therefore, to better visualise the functional data for the other data-rich exons, we scaled the Circos plot to five times the radial size as compared with the figure (figure 2) where all exons are to scale.

BRCA1 Circos including exon 11. All documented BRCA1 missense variants are represented in the Circos plot that contains 53 concentric tracks. Refer to table 1 for detailed descriptions of each track. BRCA1 Circos can be accessed via the website http://research.nhgri.nih.gov/bic/circos/ with mouse-over and search functionalities.
Interpreting BRCA1 Circos
The BRCA1 Circos plot should be read radially with track 1 being the outermost and track 53 being the innermost track. Each track is annotated by a track number and the corresponding data source (table 1). The data in each track are colour coded, and mouse-over on any data point on the figure on the website will allow the track number, track name and variant information to be displayed. As an example, we look at two variants: M1775K and M1775R (figure 3). The first track shows the exon; the variants lie in exon 21. Track 2 shows the BIC entries for each variant as a histogram (numerical values can be found in the functional database or can be seen using the mouse-over function on the website figure). In this case, there are no entries for M1775K but 31 entries in BIC for M1775R. Track 3 shows the IARC classification18 for each variant and both variants are IARC class 5 represented as a red dot. Track 4 shows the computational classification of transcriptional assay data. The classification is performed based on the posterior probability that variant is protein damaging; both these variants have a value of 1, which is represented as a histogram. Track 5 represents the Bayes’ factor and is calculated based on the transcriptional assay data; this is red for both the variants, indicating high probability of these variants being pathogenic.20 Track 6 indicates whether the variants were found in the ESP Exome Viewer. Both variants are indicated as white because they were not found in the ESP Exome viewer. Tracks 7–9 show the variant M being the reference amino acid, 1775 being the amino acid position and R and K being the variant amino acids, respectively. Tracks 11–53 show the functional impact of the variants using various functional assays and M1775K has lesser data than M1775R as shown by more white data points for M1775K. The majority of the functional assays show M1775R as having a functional impact on the protein. All the colour-coding information is shown in the database and in the track descriptions on the website for a consolidated viewing of all the information described here.

{kind=link}
{kind=link}
{kind=link}
Zooming in on an individual variant. A sample section of the BRCA1 Circos representing two missense variants, M1775K and M1775R, is shown. Each variant should be read radially to get information on all the tracks for the each variant. Refer to table 1 for detailed descriptions of each track. BIC, Breast Cancer Information Core; GVGD, Grantham Variation-Grantham Deviation; IARC, International Agency for Research on Cancer.
Functional overlap
One of the goals of the BRCA1 Circos is to help in the meta-analysis of results from individual variants. For example, a variant ‘A’ that was tested multiple times and scored as of ‘no functional impact’ in every assay is considered to have stronger evidence against functional impact than a variant ‘B’ with only one assay reported in which it scored as no functional impact. But if all the assays performed for ‘A’ are functionally redundant (they assay the exact same function) and were performed in similar conditions, then the evidence against functional impact for ‘A’ is not stronger than the evidence for ‘B’.
Data in tracks 11–53 represent results from several functional assays. Some tracks will represent data from the exact same assay reported in two or more separate papers. Others represent the same assays with differences in experimental conditions or design (eg, the transcriptional assay can be done in different cell types using the same constructs or in the same cell type but using constructs that span a different region of the protein). Finally, others can represent completely different experimental set up and assay a different function (eg, transcriptional activity in mammalian cells vs recombination in yeast). Thus, the user is cautioned to be aware of the functional overlap between assays when interpreting BRCA1 Circos data.
Website development
Using the Circos program, we generated html image maps that represent the co-ordinates of every data point on the BRCA1 Circos image. The BRCA1 Circos website (http://research.nhgri.nih.gov/bic/circos/) also displays a searchable database with information on the functional assays and functional prediction algorithms for BRCA1 variants. The functionalities of the website are
-
mouse-over ‘magnifying glass’ to zoom into any region of the Circos plot to visualise the data in that region;
-
mouse-over on every data point on the tracks shows a label with ‘track number, variant (amino acid change), track name and base change’;
-
track descriptions and key to interpret the functional data on every track;
-
all tracks hyperlinked to the original article where the data were extracted from;
-
double-clicking any part of the figure zooms into that region and allows the user to pan through the image;
-
every data point in the Circos figure is hyperlinked to the information in the BRCA1 Missense variants functional database (http://research.nhgri.nih.gov/bic/circos/allvariants/).
All codes to develop the BRCA1 Circos and raw files are available on request. In the BRCA1 Circos, each track represents one set of functional assays or prediction algorithms that indicate the functional classification of a specific BRCA1 missense variant. Two papers reporting the same assay would be displayed in two separate tracks. Two separate assays reported in the same paper would also be displayed as two separate tracks.
The current version of BRCA1 Circos depicts a specific functional assay per track. As more assays are published, the BRCA1 Circos can be updated by increasing the radius of a Circos plot which allows for more tracks to be incorporated; however, this may become unwieldy as the tool needs to fit in a standard computer screen. There are several options to solve to address this problem when it arises: (a) adding multiple BRCA1 Circos plots is a simple but the least preferable solution because we would lose the comprehensive view in one image; (b) taking advantage of our interactive interface (zooming in and mouse-over functions) and hide or show tracks (assays/studies) according to the user's choice and, finally, (c) tracks that have few variants (eg, <5) would be removed from the plot and the data would be available on the database but would not be depicted in the BRCA1 Circos.
Discussion
Mutations that disrupt BRCA1 lead to a substantial increase in risk of breast and ovarian cancers.2 ,5 Thus, genetic testing for BRCA1 is critical to identify individuals at high risk and can be used to inform clinical decisions. For example, mutation carriers might consider prophylactic surgery or increased surveillance. Current testing for BRCA1 is primarily based on sequencing exons and exon–intron borders,7 but other approaches that sequence non-coding regions such as introns and 5′ and 3′ untranslated region are starting to be used.50
Sequence-based methods can identify DNA variations that can be used to infer the outcome on the protein product. Thus, variants that cause premature or incorrect termination of the protein can, for the purposes of clinical management, be inferred to be pathogenic. However, DNA variants whose effect on the protein cannot be easily inferred from changes in the genetic code present an enormous clinical challenge.16 These variants are usually called VUS and their number is bound to increase with wide spread sequencing of clinical samples. These VUS are primarily missense variants that replace one amino acid for another and variants that may affect splicing of the transcript.
Due to the relative rarity of individual variants in the population, classical approaches such as family studies or case–control studies are difficult to conduct. Thus, in the past few years, several methods have been developed to interrogate the potential functional impact of a certain variant on the function of BRCA1.8 These methods are not without their caveats and caution is warranted when interpreting their results because BRCA1 is a multifunctional protein but it is still unclear which functions contribute to its tumour-suppressive actions. Nevertheless, results from functional assay show a strong correlation with pathogenicity and provide an important source of information to help in the classification of VUS.26 Recently, a computational model, called VarCall, was developed to convert results of functional assays into likelihood ratios of pathogenicity, allowing for functional data to be incorporated into multifactorial models of variant classification.20 Thus, we believe that using functional information will improve and accelerate the classification of variants for BRCA1.
To facilitate the process of analysing functional data, we developed a database and a visualisation tool that can display all the available functional data for individual BRCA1 variants. This tool allows for the direct comparison of results from different functional tests and provides a global picture of the functional coverage in BRCA1. It also provides a resource in which non-specialists can navigate around different functional assays and weigh information from several sources. There have been multiple efforts to facilitate systematic integration of all the data available on these variants such as the BIC (http://research.nhgri.nih.gov/bic/) and the LOVD BRCA1/2 literature (http://chromium.liacs.nl/LOVD2/cancer/home.php) database. However, there is no comprehensive resource to help interpret and visualise data available on these variants in the current literature along with the information from multiple databases. We have developed a novel web-based resource for the interpretation and visualisation of BRCA1 missense variants.
BRCA1 Circos is the first interactive tool that combines data for all documented BRCA1 missense variants with an easy-to-interpret visualisation structure. It combines information from multiple functional assays and databases in one comprehensive layout. Besides the detailed information on every variant, this tool is also useful to obtain a global view at the current data available for missense variants of BRCA1 to guide future efforts on VUS characterisation.
The data in the BRCA1 missense variant database and, consequently, BRCA1 Circos use controlled vocabulary as discussed by Lindor et al16 classifying the variants as having a ‘functional impact’, ‘no functional impact’ or ‘mild/intermediate functional impact’. This makes it easier to consolidate data from multiple studies regardless of whether the results were quantitative or qualitative. These data have been colour coded and represented in an easy-to-use manner with many different features such as zooming into a particular exon, variant and obtaining information on multiple variants at a glance. We also provide the original articles for every track in the Circos and it is advisable to read the original publications to obtain any experimental data.
Several limitations apply to the data in the BRCA1 Circos. In particular, because most assays described here have not been validated extensively nor were conducted with specific standard operating procedures in a clinical environment, results should be taken with caution. In addition, individual-level data could suffer from clerical errors, swapped samples, mistakes in constructs and other problems that are inherent to research.13 As the gold standard for VUS classification14–16 requires that more than one independent source of data to classify a variant, functional data represented in the BRCA1 Circos should not, at the present stage, be considered as a single source for the purposes of clinical decisions. It is expected that as assays are validated and reported as likelihood ratios (eg, as in VarCall), the functional data will be incorporated in the multifactorial model. For rare variants for which genetic data may not be available, it is possible that properly validated assays could be used as a single source for clinical decisions but this approach needs to be further assessed in future studies.
In conclusion, we have developed a web-based tool to visualise complex data on BRCA1 variants using an open-source resource—Circos.21 This study can be used as a platform for the development of a comprehensive functional database for other cancer susceptibility genes such as BRCA2 and can be used by researchers and genetic counsellors to better interpret the function of VUS.
Acknowledgments
The authors would like to sincerely thank Tyra Wolfsberg and Suiyuan Zhang for helping with webpage development and all individuals and families who have generously donated their time, samples and information to facilitate research on the predisposition factors of breast and ovarian cancer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online table 2
- Data supplement 3 - Online table 3
Footnotes
-
Collaborators ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) Consortium.
-
Contributors AJ, AV and ANM conceived the project. AV, RSC, GAM, MV and SVT contributed to the development of the database. AJ designed and developed the Circos images. AJ, RCJ and BK designed and developed the web tool. PW, ABS, MPGV, SMC, GAM, AV, NC, AG, DE, MJB, TP, RBvdL, MSP, SLN, TD, EM, ST, MV, FJC, SVT, JNMG, MAC, LCB and SKS contributed to the curation of the database and the testing and refinement of the web tool. AJ, AV and ANM wrote the paper. All authors contributed to the discussion and overall data interpretation and approved the final manuscript.
-
Funding This work was funded by H. Lee Moffitt Cancer Center Foundation (Miles for Moffitt), and the US National Cancer Institute. The ENIGMA consortium is funded by a supplement to NIH RO1 award CA116167.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All data will be available for public use and download at the website for the BRCA1 Circos (NHGRI).