Article Text

Abstract
Background Neuromuscular disorders are a clinically, pathologically, and genetically heterogeneous group. Even for the experienced clinician, an accurate diagnosis is often challenging due to the complexity of these disorders. Here, we investigated the utility of next generation sequencing (NGS) in early diagnostic algorithms to improve the diagnosis for patients currently lacking precise molecular characterisation, particularly for hereditary myopathies.
Methods 43 patients presenting with early onset neuromuscular disorders from unknown genetic origin were tested by NGS for 579 nuclear genes associated with myopathy.
Results In 21 of the 43 patients, we identified the definite genetic causes (48.8%). Additionally, likely pathogenic variants were identified in seven cases and variants of uncertain significance (VUS) were suspected in four cases. In total, 19 novel and 15 known pathogenic variants in 17 genes were identified in 32 patients. Collagen VI related myopathy was the most prevalent type in our cohort. The utility of NGS was highlighted in three cases with congenital myasthenia syndrome, as early diagnosis is important for effective treatment.
Conclusions A targeted NGS can offer cost effective, safe and fairly rapid turnaround time, which can improve quality of care for patients with early onset myopathies and muscular dystrophies; in particular, collagen VI related myopathy and congenital myasthenia syndromes. Nevertheless, a substantial number of patients remained without molecular diagnosis in our cohort. This may be due to the intrinsic limitation of detection for some types of mutations by NGS or to the fact that other causative genes for neuromuscular disorders are yet to be identified.
- Peripheral nerve disease
- Motor neurone disease
- Molecular genetics
- Muscle disease
- Neuromuscular disease
Statistics from Altmetric.com
Introduction
Neuromuscular disorders (NMDs) are genetically heterogeneous muscle diseases with several different types, varying age of onset, clinical severities, and broad pathologic findings. Particularly in children there are non-specific clinical features such as motor developmental delay, hypotonia, and overt weakness, which make it difficult to reach a specific diagnosis even for the experienced clinician.1–3 The prevalence of congenital myopathies (CMs) is estimated at about 1:25 000.4–7 The prevalence of congenital muscular dystrophy (CMD) is not well known; the relative frequency of each subtype appears to be variable in different countries.8–10 Given the diagnostic difficulties, the variable clinical spectrum and the high rate of de novo events, the overall prevalence of NMDs may be underestimated.
While for some hereditary myopathies such as Duchenne/Becker muscular dystrophy, the initial diagnostic step is a single molecular genetic test, for many patients with highly suspected myopathies a muscle biopsy is still required. This procedure is particularly challenging in newborns, infants, or young children. Moreover, substantial numbers of patients remain undiagnosed even after extensive pathologic studies, due to the lack of specific markers for many myopathies or to ambiguous and inconclusive results. Furthermore, Sanger sequencing of individual genes known to be associated with NMDs, in particular hereditary myopathies, is time consuming, cost prohibitive, and cannot be prioritised due to heterogeneous genetic backgrounds and similar clinical presentations. One gene can cause a wide variety of clinical and/or pathological features, while similar clinical features can be caused by mutations in different genes. For example, nemaline myopathy can be caused by defects in at least eight different nuclear genes. Mutations in one of those eight genes, ACTA1, can cause different pathological findings, such as central core disease or congenital fibre type disproportion. In addition to the high degree of genetic heterogeneity, the large size of the genes such as RYR1 with 106 exons, or TTN with 362 exons, is a significant barrier for a time and cost effective molecular genetic diagnosis.
Recent studies on the clinical application of next generation sequencing (NGS) technology have started a new era of molecular genetic diagnosis. We also utilised the NGS for the diagnosis of mitochondrial disorders.11–13 Both targeted and whole exome NGS sequencing approaches have been tested for the molecular diagnosis of CMD and congenital myopathies in a small subset of patients.14 ,15
Hereditary NMDs encompass a significant proportion of patients with chronic muscle disease. Accurate molecular genetic diagnosis can improve management, provides appropriate counselling, and allows for potential therapeutic trials.
Here, we present the results of an NGS panel test targeting 579 genes on 43 patients with early onset NMDs. All of these patients lacked a definite molecular diagnosis despite previous muscle biopsies and/or multiple gene sequencing tests. Our goal was to establish the clinical usefulness of targeted NGS in NMDs with early presentation.
Methods
Patients
The clinical database at Seoul National University Children's Hospital was reviewed and 43 patients presenting with early onset (<5 years) hypotonia and/or muscle weakness of unknown genetic origin were selected for this study. Hypotonia due to central nervous system (CNS) or chromosomal abnormalities were excluded. All of the patients were Korean. Informed consent was obtained for the collection of clinical data and extraction of DNA to perform genetic analysis. The institutional review board (IRB) of the Seoul National University Hospital approved the study protocol. The NGS study at Seattle Children's Hospital Research Institute was exempted from approval by the IRB at the institute. Before the NGS testing, genetic tests including spinal muscular atrophy, Duchenne muscular dystrophy, MTM1, and myotonic dystrophy were performed, based on the clinical and pathologic findings in some patients, but were negative. Most of the patients had clinical diagnoses of congenital myopathy, muscular dystrophy or unknown NMDs. Histopathological phenotype was reviewed in 40 cases that underwent muscle biopsies at Seoul National University Children's Hospital. Serial frozen sections from each muscle sample were stained using a set of histochemical methods including haematoxylin, modified Gomori trichrome, nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR), succinyl dehydrogenase, ATPase, and immunohistochemistry for dystrophin, merosin and dysferlin. Immunohistochemical staining with an anti-collagen VI α antibody (Chemicon, #MAB1944) was performed on clinically suspected patients with collagenopathies.
Targeted genes and sample preparation library
A DNA library was prepared for each sample by capturing the exons of the genes of interest using custom made DNA probes (Haloplex Agilent, Santa Clara, California, USA). The selected genes (see online supplementary table S1) comprised 10,706 exons and a total of 3.88 Mb. These were genes for various NMD including: congenital muscular dystrophies, congenital myopathies, metabolic and mitochondrial myopathies, storage myopathies, distal myopathies, channelopathies, disease of neuromuscular junction and peripheral nerve. The list also included 199 genes selected from mitochondrial components, because mutations in these proteins often manifest as myopathies. Some of the genes were considered as candidate genes that have not been reported as pathogenic yet, but belong to functional groups that are usually pathogenic.
Sequencing analysis
Forty-three samples were sequenced at 5–8 samples per lane on a GAIIx instrument (Illumina, San Diego, California, USA) using 2×100 paired-ends reads. Reads were aligned using Burrows-Wheeler Aligner (BWA, V.0.7.3) and data were analysed with the Genome Analysis Toolkit (GATK, V.2.4.9) (Broad Institute, Cambridge, Massachusetts, USA) as previously described.13
Variant annotation and identification of mutations
Variants were annotated using SeattleSeq16 and wAnnovar.17 Variants found within the targeted regions were further evaluated for their possible clinical significance by cross-referencing to the Single Nucleotide Polymorphism Database (dbSNP), the Exome variant server, and the 1000 Genomes browser. An internal database of polymorphisms was also used for quality assurance during this analysis. PolyPhen-2 and SIFT (sorting intolerant from tolerant) scores were used only for reference but were not used for filtering the variants. For variants in genes with autosomal dominant and X-linked disease inheritance, we used the minor allele frequency (MAF) cut-off of 0.2%, and for variants in genes with autosomal recessive disease inheritance, we used the MAF cut-off of 0.5%.13 Variants that exceeded these frequencies were not considered as potential mutations, even if they were previously reported as such in the Human Gene Mutation Database (HGMD). Finally, variants were searched in the HGMD by Genometrax.18 ,19 The sequence variants were interpreted based on the guideline from American College of Medical Genetics.20
Confirmation
Variants identified as possibly disease causing were confirmed by standard PCR combined with Sanger sequencing using a Big-Dye Terminator v3.1 kit on an ABI3130xl automatic DNA sequencer system (Applied Biosystems, Carlsbad, California, USA). Segregation of variants in the family was assessed by sequencing parental DNA samples when available.
Results
Sequencing summary
The sequencing yield was on average 1.6 Gb per sample. On average 74% of the reads mapped to the intended targets and 96% of the targets had at least 20 reads per base, with an average of 175 reads per base. On average about 300 exons tended to have <20 reads with an average GC content of 57%. Approximately 120 variants per sample were detected in exons or splice sites that were not present in the dbSNP or in the exome server (on average, 67 missense, four in splice sites, one frameshift, and 51 coding synonymous).
Spectrum of genes with suspected variants
In 32 out of 43 patients tested, we detected 19 novel variants, 15 known pathogenic variants, and one variant previously reported as variant of uncertain significance (VUS) in 17 genes associated with CMD, congenital myopathy, congenital myasthenia, cardiomyopathy, and metabolic disease (figure 1). COL6A1-3 and RYR1 genes were most frequently affected in our cohort. The number of cases and distribution are illustrated in figure 1.

Distribution of pathogenic, likely pathogenic and variants of uncertain significance (VUS) variants by the type of neuromuscular disorders. The number of cases are listed next to the gene names.
Identification of pathogenic mutations with phenotypic characteristics
Pathogenic mutations were confirmed in 21 cases (48.8%) out of 43 patients with an early onset NMD (table 1).
Pathogenic variants, likely pathogenic variants and variants of uncertain significance in 32 patients
Muscular dystrophies
Eleven patients were diagnosed with muscular dystrophies. COL6A mutations were identified in eight cases having clinical phenotypes of Ullrich congenital muscular dystrophy (UCMD) or Bethlem myopathy (BM). Various degrees of sarcolemma-specific collagen deficiency in collagen VI α immunostaining were observed in muscle biopsy sections from all eight patients. Six of the mutations were de novo and the other two were inherited from one of the parents, who were also affected (case 8 and 9).
All three cases (6, 16, 55) with LMNA gene defects had de novo mutations previously reported.
Congenital myopathies
Five cases were genetically diagnosed with congenital myopathies. Two nemaline myopathy patients were confirmed to have de novo mutations in the ACTA1 gene. Case 24 was a hemizygote for a missense variant inherited from the mother in the MTM1 gene and case 7 carried two variants in trans in the RYR1 gene. Both patients shared typical clinical features of congenital myopathy, such as very early onset muscle weakness, hypotonia, high arched palate, and normal creatine kinase (CK) concentration.
Congenital myasthenia
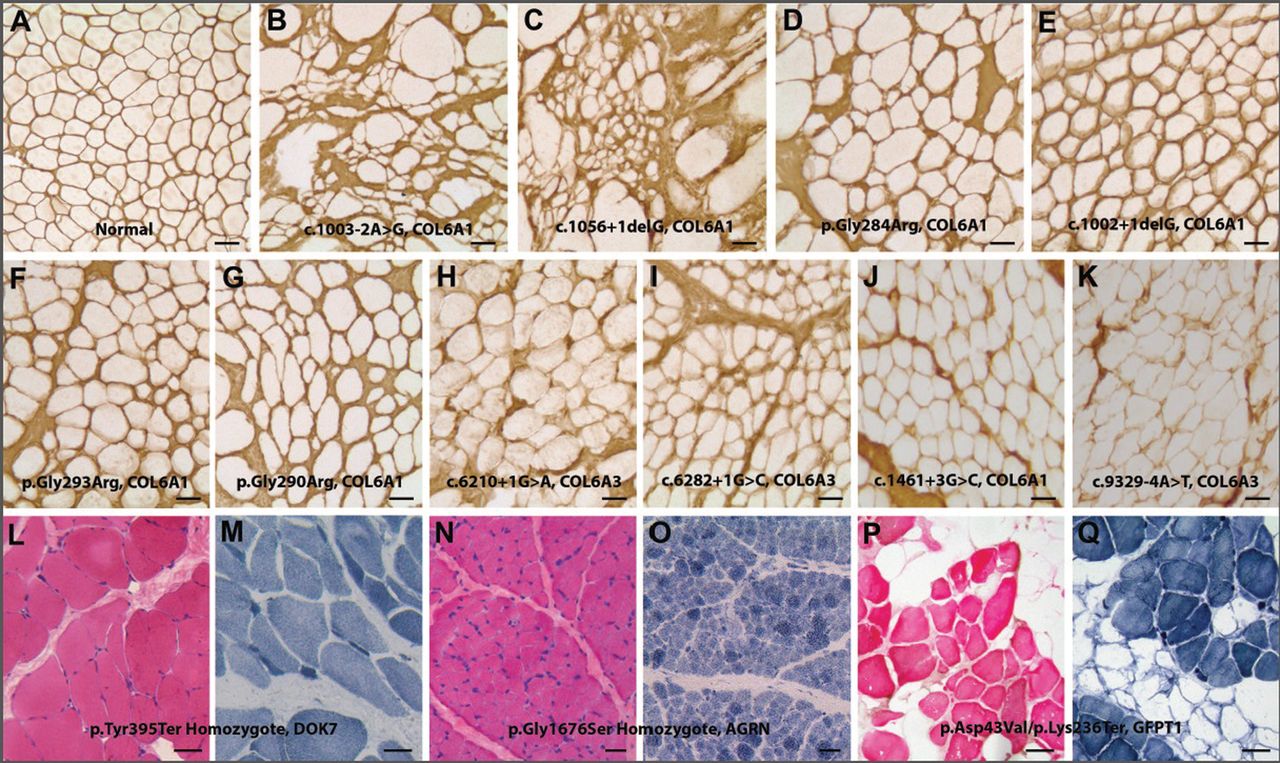
Three patients with congenital myasthenia were identified. Their provisional diagnosis after muscle biopsy was congenital myopathies (cases 5 and 30) and limb girdle type muscular dystrophy (case 11). All of these patients showed various myopathic phenotypes with early onset muscle weakness, respiratory failure, and very early onset scoliosis. Muscle biopsy revealed non-diagnostic findings such as fibre size variations and/or endomysial fibrosis (figure 2L–O) or some degenerative fibres with fatty infiltration (figure 2P, Q).
{kind=link}
{kind=link}
Muscle pathology findings. (A–K) Collagen VI α immunostaining images of muscle biopsy from 10 patients with pathogenic/likely pathogenic COL6A1 and COL6A3 mutations. Various degrees of sarcolemma-specific collagen deficiency are noted (stain with anti-collagen VI α antibody and horseradish peroxidase-DAB). (A) Normal control, (B) case 8, (C) case 9, (D) case 10, (E) case 22, (F) case 31, (G) case 41, (H) case 46, (I) case 52, (J) case 29 and (K) case 48. (L–Q) Haematoxylin and eosin (left) and NADH-tetrazolium reductase (TR) stains (right) of muscle sections from three patients with congenital myasthenic syndrome. (L and M) Case 5—DOK7 mutation, (N and O) case 30—AGRN mutation, (P and Q) case 11—GFPT mutations. Bar denotes 50 µm.
A homozygote known nonsense mutation in the DOK7 gene was found in case 5. In case 11, a known missense and a novel nonsense variant were detected in trans in the GFPT1 gene. Case 30 was homozygous for a novel missense variant in the AGRN gene.
Neuropathies
Mutations in genes related to neuropathy were identified in two patients. Case 13 carried a novel, de novo alteration in the GARS gene. This patient presented with severe hypotonia since birth and had a negative SMN gene test. Case 28, who had presented with hypotonia since birth, carried a novel, de novo alteration in the DYNC1H gene. A femur fracture was also identified at birth.
Likely pathogenic variants and VUS
In two patients, the clinical and immunohistological phenotype supported the classifications of two novel variants as likely pathogenic: case 29 (figure 2J) with a variant in COL6A1 gene, inherited from asymptomatic father, and case 48 (figure 2K) with a variant in COL6A3 gene. In six patients, we identified likely pathogenic variants in RYR1, CCDC78, COL6A1, COL6A3, MYBP3 and DNA2 genes (table 1). Their phenotypes were consistent with previously described phenotypes associated with each gene. Of note, in three patients (cases 3, 29 and 37), their dominant variant alleles were inherited from one of their asymptomatic parents.
Nucleotide variants in RYR1, DCTN1, and COL6A2 were detected in four patients with clinical phenotypes consistent with previously described presentations. However, we classified these novel variants as VUS because parental testing was not available (table 1).
No suspected variants were identified in 11 patients (see online supplemental table S2).
Discussion
Our study illustrates the clinical benefit of targeted NGS in diagnosing patients with early presentation of NMDs of undetermined molecular aetiology. While whole exome sequencing is being clinically used, it is more costly, has a higher false positive rate, a longer turnaround time, and is more difficult to interpret, compared to targeted NGS. Targeted NGS for NMD testing is also less invasive and presumably more cost effective and time saving than the conventional approaches which require a muscle biopsy and Sanger sequencing of several suspected genes. We designed the NGS test to include all genes implicated in NMDs, focusing in particular on hereditary myopathies. This choice was based on the observation that both clinical and laboratory diagnosis of heterogeneous genetic conditions, even if performed by experienced physicians and pathologists, is often extremely challenging due to overlapping clinical phenotypes, expanded clinical spectrums, difficulties to obtain a muscle biopsy, and the complexity of performing and interpreting immunohistochemical stains.12 ,13
The overall detection rate in our patient cohort was fairly high (48.8% with definitely pathogenic mutation). When likely pathogenic and VUS variants were included, it was 76.2% (32 over 43 patients). The classification of the genes is illustrated in figure 1. We believe that this high rate is due to the fact that the patient cohort was carefully selected by specialised muscle experts and represented a fairly homogeneous group. Interestingly, 15 of the observed variants in this study were recurring mutations previously reported, and 13 patients had a de novo variant.
Our study confirms that mutations in the genes for the extracellular matrix protein collagen VI α are a prevalent cause of CMD, with a spectrum of presentations ranging from BM to UCMD.8 ,14 ,21 Case 52, with merosin positive CMD and suspected limb girdle muscular dystrophy (LGMD), actually had a de novo variant of COL6A3 predicted to abolish a donor splice site function. The pathogenicity of this variant was further supported by abnormal immunohistochemical staining (figure 2I). Collagen VI related contractures may be relatively subtle, leading to potential confusion in diagnosis with cases of LGMD.22 This case indicates that the spectrum of collagenopathy could be broader than we thought and some patients could be potentially misdiagnosed. Three patients (figure 2D, F, G) carried known mutations in the triple helical domain in glycine residues previously reported in patients affected by UCMD and BM.23–25 Variable clinical phenotypes have been previously observed in the cases with glycine substitutions.24 Some cases can present with subtle abnormalities or even with normal immunohistochemical staining.26 It was noted that the immunohistochemical findings were marginally abnormal in these three cases while their clinical phenotypes were consistent with early onset UCMD. Considering the occasional tricky interpretation of collagen VI expression in some biopsies and the fairly large size of the involved three genes, the application of NGS as an early diagnostic step seems reasonable and could be beneficial. In fact, it may provide critical genetic information that can help predict the outcome, and that the risk associated with general anaesthesia and invasive procedure can be reduced.
All three cases with LMNA gene defects had de novo mutations previously reported as pathogenic. Their provisional diagnosis before this study was CMD, unknown type. LMNA related myopathies are well known to have variable clinical phenotypes with variable severities and pathologic findings, ranging from early onset severe myopathies to late onset limb girdle muscular dystrophies. Therefore, in clinical practice, a specific diagnosis is often difficult even for the experienced clinician. In our cohort, case 6, who showed a mild dystrophic pattern in pathology, carried a known mutation previously described in a severe CMD patient who died of respiratory and heart failure.27 ,28 This highlights the extremely variable clinical presentations even from the same mutation. Case 16 and 55 carried known mutations consistent with the patients’ laminopathy phenotype. The early diagnosis of LMNA-associated myopathy is particularly important because the patients eventually develop severe cardiac problems, with high mortality,29 ,30 and can benefit from prophylactic implantable defibrillator therapy.31 ,32
As in other studies of similar patients, mutations in ACTA1 and RYR1 were common causes of congenital myopathy in our cohort.7 Two cases with RYR1 gene variants (7 and 57) carried two variants in trans while the other three were heterozygotes. Mutations in the RYR1 gene are associated with malignant hyperthermia, central core disease, minicore myopathy with external ophthalmoplegia, and congenital myopathy. Muscular dystrophy is also observed for RYR1 mutations, and this was the case for one patient who was a compound heterozygote for a known and a novel likely pathogenic mutation (case 57) (table 1). This case was classified as muscular dystrophy, given the pathological features, while the other four cases presented with congenital myopathy. Case 37 had a known alteration in the RYR1 gene and was classified as likely pathogenic based on his clinical and pathological features, although his mother carried the same allele and was asymptomatic. This mutation, p.Phe492Ser, is located in hotspot domain 3 and was previously reported as dominantly acting in a patient affected by central core disease.33
Along with this case, a few other cases (case 29—COL6A1 gene, case 3—MYBPC3, and case 4—CCDC78) carried suspected variants inherited from their asymptomatic parents, while the clinical or muscle pathology findings supported the suspected molecular defects, which imply that incomplete penetrance, variable expression in the same families, or mosaicism may be possible. This speculation was further supported in case 29 by abnormal collagen VI staining in muscle pathology, while his father with the same variant has been asymptomatic to date (figure 2J). While the father appeared to have the same load of mutant allele in DNA extracted from blood (Sanger data, not shown), it is possible that a lower load in muscle tissue due to mosaicism may explain the differential presentation. Therefore, a careful interpretation is required for the analysis of cosegregation in the family, as many hereditary myopathies are autosomal dominant in inheritance. The parental conundrum, due to possible variable penetrance or mosaicism, has been described for collagen VI mutations22 ,24 ,34 but not for RYR1, MYBPC3 or CCDC78 gene mutations yet.
It is noteworthy that six patients, who were previously suspected to have either muscular dystrophy or congenital myopathy, were found to have congenital myasthenic syndrome (CMS), motor neuron disease or peripheral nerve disease. These types of conditions are difficult to diagnose by pathologic findings (table 1 and figure 2L–Q) and symptoms are often non-specific. Particularly in CMS, early diagnosis is very important since treatment with acetylcholinesterase inhibitors, salbutamol or ephedrine, can be effective, and patients can ultimately have a normal life expectancy.35 In fact, after the molecular genetic diagnosis of CMS in our three patients was made, we started the treatment in two patients. In case 5, motor performance was much improved after 3 months treatment with salbutamol, such that the patient can now stand up and walk independently.
In case 28, a de novo missense variant in gene DYNC1H was found, affecting a residue of the dimerisation domain. DYNC1H1 codes for a cytoplasmic dynein heavy chain that is involved in retrograde transport along microtubules. Mutations in DYNC1H1 have been associated with autosomal dominant spinal muscular atrophy and Charcot–Marie–Tooth disease.36 Moreover mutations have also been associated with malformations of cortical development37 and intellectual disability.38 Our case may indicate an even broader clinical spectrum associated with this gene defect.
In case 11 with GFPT1 gene mutations, some of the clinical features of proximal dominant muscle weakness of early onset were similar to previously reported patients.39 However, dystrophic findings in pathology without tubular aggregates, along with facial weakness and ptosis, broaden the spectrum of GFPT1 related myasthenic syndrome. The missense variant in the glutaminase domain was previously described in a patient with a limb-girdle pattern of muscle weakness combined with the presence of tubular aggregates on muscle biopsies.40 The nonsense variant is in the alternative cassette exon of transcript variant 1, which is specifically expressed in muscle.41 Disruption of muscle-specific isoform has been associated with devastating clinical phenotypes.39 We started acetylcholine esterase inhibitor treatment and have been following his motor performance for over 3 months. The patient has shown a significant improvement in motor strength but still cannot walk alone. All these cases highlight the utility of targeted NGS for the clinical diagnosis of muscle weakness, which has quite a broad range of aetiology overlapping with many types of myopathies.
In summary, targeted NGS in conjunction with clinical and pathological findings can help reach a precise molecular genetic diagnosis in early onset NMDs, in particular hereditary myopathies. Targeted NGS could be advantageous when applied in an early diagnostic algorithm for early onset NMDs to avoid invasive procedures and risk associated with general anaesthesia. However, careful interpretation is necessary especially for possible incomplete penetrance, mosaicism or variable expression for certain cases. Understanding the specific molecular defects can help provide appropriate tailored management with potential significant improvement in prognosis.
Acknowledgments
We thank Ms Thao Tran for her technical assistance. We thank to Dr Margaret Sedensky for a critical editing on our manuscript. We deeply thank our patients and families who participated in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
Footnotes
-
JHC and VV contributed equally.
-
Contributors JHC: participated in design and conceptualisation of this study, analysis and interpretation of the data and drafting the manuscript. VV: participated in design and conceptualisation of this study, performed the experiments, participated in analysis and interpretation of the data and drafting the manuscript as equally as JHC. AC: participated in analysis and interpretation of the data and drafting the manuscript. BCL: participated in analysis and interpretation of the data. SHE: participated in analysis and interpretation of the data. QZ: participated in analysis and interpretation of the data. SHH: as a PI designed and conceptualised this study, analysed and interpreted the data and drafted the manuscript.
-
Funding This study was partly supported by the funding awarded to VV from Northwest Mitochondrial Research Guild. JHC was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (Grant No. HI13C1468).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Seoul National University, Seattle Children's Hospital.
-
Provenance and peer review Not commissioned; externally peer reviewed.