Article Text

Abstract
Introduction Kinesin superfamily (KIF) genes encode motor proteins that have fundamental roles in brain functioning, development, survival and plasticity by regulating the transport of cargo along microtubules within axons, dendrites and synapses. Mouse knockout studies support these important functions in the nervous system. The role of KIF genes in intellectual disability (ID) has so far received limited attention, although previous studies have suggested that many ID genes impinge on synaptic function.
Methods By applying next-generation sequencing (NGS) in ID patients, we identified likely pathogenic mutations in KIF4A and KIF5C. To further confirm the pathogenicity of these mutations, we performed functional studies at the level of synaptic function in primary rat hippocampal neurons.
Results and conclusions Four males from a single family with a disruptive mutation in the X-linked KIF4A (c.1489-8_1490delins10; p.?- exon skipping) showed mild to moderate ID and epilepsy. A female patient with a de novo missense mutation in KIF5C (c.11465A>C; p.(Glu237Lys)) presented with severe ID, epilepsy, microcephaly and cortical malformation. Knock-down of Kif4a in rat primary hippocampal neurons altered the balance between excitatory and inhibitory synaptic transmission, whereas the mutation in Kif5c affected its protein function at excitatory synapses. Our results suggest that mutations in KIF4A and KIF5C cause ID by tipping the balance between excitatory and inhibitory synaptic excitability.
- KIF4A
- KIF5C
- intellectual disability
- cortical malformation
- synaptic function
Statistics from Altmetric.com
Introduction
Kinesin superfamily proteins (KIF) are motor proteins involved in the movement of various cargoes along the microtubules, including vesicles, organelles, protein complexes, mRNAs and chromosomes.1–4 KIF proteins act together with motor proteins from the dynein and myosin superfamilies. These molecular motors are expected to have fundamental roles in several processes in the brain, including neuronal functioning, development, survival and plasticity, by regulating the anterograde and retrograde transport within the axons, dendrites and synapses of neurons.1–4 Studies in mouse models support these essential functions of KIF genes in the development and functioning of the nervous system. Mice with homozygous knockout mutations in Kif1a, 1b, 2a, 3a, 3b, 4a, 5a and 5b show various neurological phenotypes including structural brain anomalies, decreased brain size, loss of neurons, reduced rate of neuronal apoptosis and perinatal lethality due to neurological problems.4–12 The embryonic lethality of knockout mice for Kif2a, Kif3a and 3b, and Kif5b suggest that these Kif genes have an important function in general developmental processes as well.5 ,7 ,8 ,13 Overexpression of Kif17 in mice resulted in enhanced spatial learning.14 Conditional knockout of KIF5A in mice led to epileptic phenotypes.15
Several KIF genes have previously been implicated in the pathogenesis of neurodegenerative and neurodevelopmental disorders in humans (table 1).
Kinesin superfamily (KIF) genes implicated in neurodegenerative and neurodevelopmental diseases in humans
Only a handful of previous reports relate to mutations in KIF genes to intellectual disability (ID). Putoux et al21 identified homozygous mutations in KIF7 (MIM 611254) in patients with acrocallosal syndrome (MIM 200990) and hydrolethalus syndrome (MIM 614120), and homozygous mutations in the same gene were also found in patients with Joubert syndrome 12 (MIM 200990).22 In these ciliary disorders ID is part of the phenotype. Najmabadi et al23 identified a homozygous missense mutation in KIF7 by targeted sequencing of exons from homozygous linkage intervals in a consanguineous family with ID, clubfeet, cataract, hearing loss and midface hypoplasia. More recently, the first dominant de novo missense mutation in a KIF gene (KIF1A) was found in a patient with non-syndromic ID.17 Recently, Poirier et al19 reported the identification of mutations in KIF2A and KIF5C in patients with malformations of cortical development (MCD). These patients presented with ID as well.
Here, we report the identification of a mutation in KIF4A (MIM 300521) in an X-linked ID family, presenting with mild to moderate ID and epilepsy, and a mutation in KIF5C (MIM 604593) in a sporadic patient with severe ID, microcephaly, epilepsy and cortical malformation. Additionally, we studied the effects on synaptic function of these KIF genes using a knockdown system in rat primary neurons.
Methods
Patient ascertainment
Family 1 was included in a larger project involving sequencing of all exons of the X-chromosome in >200 families with X-linked ID. For each family, DNA from one affected male was subjected to targeted enrichment of all X-chromosome-specific exons followed by next-generation sequencing (NGS). The enriched and sequenced exons encompassed 99.2% of the 7759 disease-causing X-chromosomal changes listed in the Human Gene Mutation Database (HGMD, update April 2011). Variants were filtered against dbSNP135, control populations (185 genomes from the 1000 Genomes Project Consortium; http://www.1000genomes.org/data, 200 Danish exomes, the NHLBI Exome Sequencing Project (ESP, freeze 5400)) as well as an in-house database (Kalscheuer et al, personal communication). Variants with a minor allele frequency of ≥0.5% were excluded.
In patient 2, we performed family based whole-exome sequencing as previously described by de Ligt et al.26
Confirmation of mutations and segregation analysis were done by Sanger sequencing.
RNA expression studies in cell-lines of patients from family 1 with a mutation in KIF4A
RNA was isolated from a cell-line from an affected family member and a healthy control person. Primers for PCR analysis were designed at exon 13/14 and exon 16/17 boundaries and cDNA was synthesised. Sequencing of the PCR product in an affected family member and the healthy control person was performed to confirm skipping of exon 15 in the patients.
siRNAs, DNA constructs, hippocampal neurons and transfection
The following validated siRNAs were obtained from Sigma Aldrich: Kif4a; SASI_Rn02_00290987 5′-CUAAUGACUUCUGUAGCUU-3′; SASI_Rn02_00290988 5′-CAUCUAGAACUGAAACCUA-3′; SASI_Rn02_00290989 5′-CAACCUAGAGGAAACAUUA-3′. Kif5c; SASI_Rn02_00231705 5′-GGUUACAAUGGGACAAUUU-3′; SASI_Rn02_00231706; 5′-GCUUCUAGCUUCCACCAGA-3′; SASI_Rn02_00231707 5′-CAGAGAACUCCAGACUCUU-3′. The BLOCK-iT fluorescent oligo (Invitrogen) that is not homologous to any known genes was used as transfection efficiency detector and a negative control. Each siRNA was tested in hippocampal neurons and validated using quantitative PCR. Primary hippocampal neurons were prepared as described previously27 and transfected with siRNAs at 6 days in vitro (DIV). Three days later, whole-cell recordings were obtained from transfected neurons. In experiments in which we examined the role of KIF5C wild type (WT) and KIF5C-E237 K on miniature excitatory postsynaptic currents (mEPSCs), primary hippocampal neurons were electroporated immediately after dissociation using the Amaxa Nucleofector device (Amaxa GmbH). 3 μg total of green fluorescent protein (GFP), KIF5C-WT and KIF5C-E237 K plasmids was used per electroporation. Whole-cell recordings were conducted from transfected neurons in every group. The YFP-KIF5c (KIF5c-WT) construct was a gift from Dr A Stephenson. The KIF5C mutant (KIF5C-E237 K) was generated by introducing a point mutation (E237 K) using the Quick Change system (Stratagene).
Measurement of mEPSCs and measurement of miniature inhibitory postsynaptic currents in rat primary hippocampal neurons
Whole-cell voltage-clamp recordings were performed from control neurons or neurons transfected with siRNAs against Kif4a or cDNA-expressing KIF5C-WT/E237 K at holding potential of −60 mV in artificial cerebro-spinal fluid (ACSF) containing 2.5 mM CaCl2 and 1.2 mM MgCl2 at 30°C. mEPSCs were recorded in the presence of 1 μM tetrodotoxine (TTX) and 0.1 mM picrotoxin. Miniature inhibitory postsynaptic currents (mIPSC) were recorded in the presence of 1 μM TTX , 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 100 μM ((2R)-amino-5-phosphonovaleric acid (APV). Five to 10 min of recordings were analysed from each cell using the Mini Analysis Program (Synaptosoft) using 8 pA as a threshold to detect an event. For each cell, we analysed a minimum of 500 events. The amplitude of mEPSCs and mIPSCs is directly related to the postsynaptic strength, whereas, the frequency is correlated to the presynaptic release properties and/or the amount of functional synapses.
Western blot analysis
Cells from control and patients were lysed in ice-cold lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 40 mM β-glycerophosphate, 2 mM Na3VO4, and complete protease inhibitors (Roche). Cell extracts in Laemmli buffer were resolved by SDS-PAGE (using mini protean TGX gels, Biorad), transferred to a nitrocellulose membrane (Trans-blot turbo transfer pack, Biorad), and analysed by immunoblotting with the indicated antibodies. The following antibodies were used: rabbit monoclonal anti-KIF4A (Ab124903, Abcam), monoclonal anti-gamma tubulin (T5326, Sigma).
Immunofluorescence
To detect the localisation of KIF5C-WT and KIF5C-E237 K transfected primary hippocampal neurons were fixed at 21 DIV in 4% paraformaldehyde with 4% sucrose solution, permeabilised with 0.2% Triton X-100 in phosphate buffered saline (PBS), and incubated overnight with the following primary antibodies: anti-MAP2 (synaptic system) and anti-GFP (Abcam). As secondary antibodies we used: Alexa-568 and Alexa-488 donkey antirabbit or antiguinea pig (Invitrogen). Immunofluorescence was visualised under a fluorescent microscope (Axio Imager Z1; Zeiss, Basle, Switzerland) equipped with a camera (AxioCam MRm; Zeiss). Images were processed using Axiovision Rel (V.4.6) and analysed using ImageJ (NIH, Bethesda, Maryland, USA). For each condition, 3 dendrites per neuron were analysed (n=6 neurons/group). Dendritic signal of KIF5C-WT and KIF5C-E237 K was normalised to its signal in the soma.
Results
Clinical description patients
Family 1
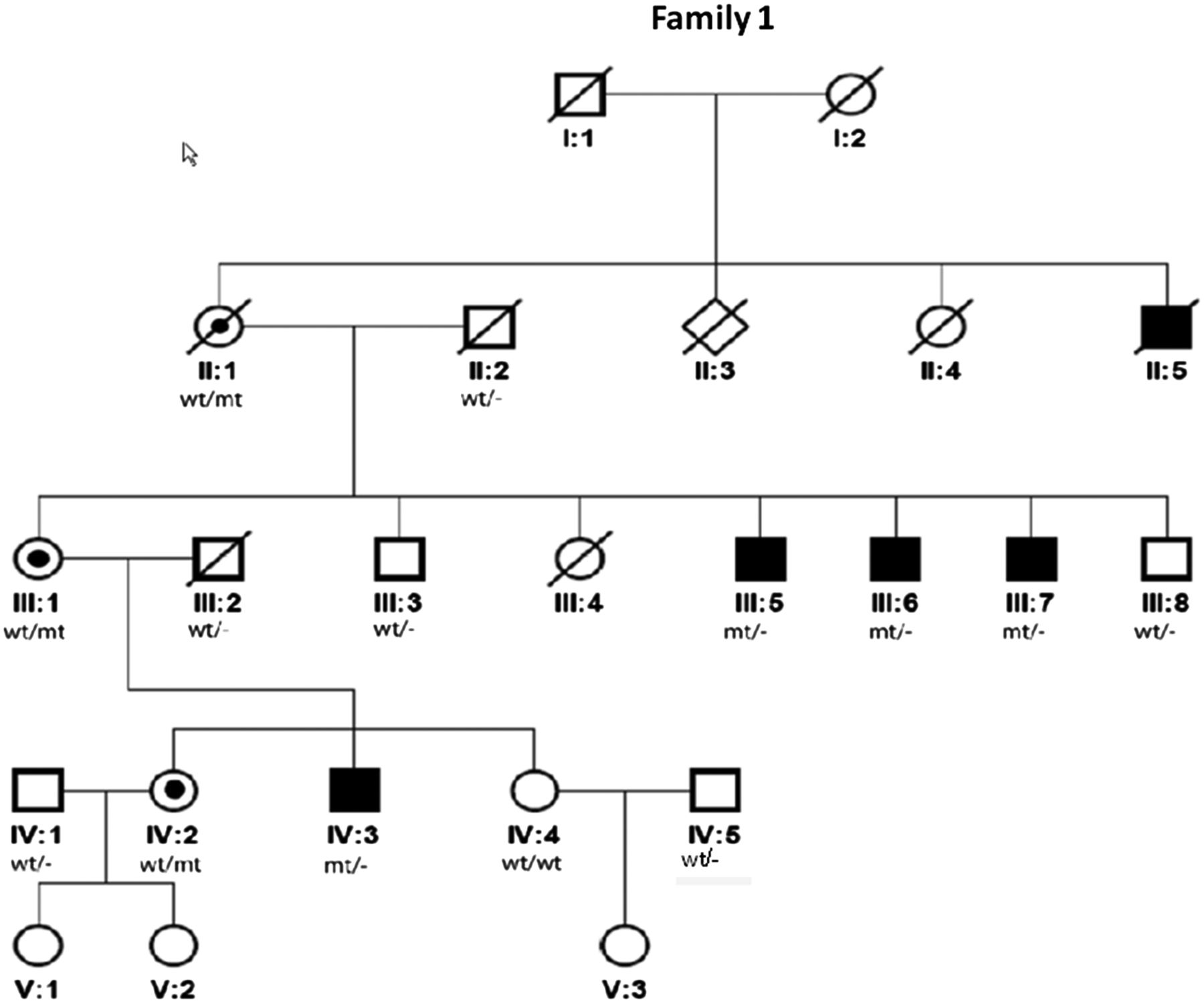
We searched for the causative mutation in a family (family 1) with five affected males in different generations (figure 1), suggesting X-linked recessive inheritance. The patients presented with mild to moderate ID, were able to speak simple sentences and lived and worked in sheltered places. Four of the five individuals had epilepsy that manifested at later childhood or adolescent age and comprised complex partial and generalised seizures (both absences and tonic-clonic seizures). Head circumferences were small to low-normal (0.6th–10th centile). Facial dysmorphisms were mild and non-specific. A CT scan of the brain in two of the probands (III:7 at age 53 years and IV:3 at age 7 years) had shown central atrophy of lateral hemispheres and wide posterior horns, respectively. Previous genetic investigations in this family included conventional karyotyping, genome-wide array analysis by the 2.7M Affymetrix array platform, DNA analysis of FMR1 (MIM 309550) and ARX (MIM 300382), and a metabolic screen. The results did not provide an explanation for their phenotype. Linkage analysis, based on an X-linked recessive inheritance pattern with complete penetrance, indicated two intervals with a LOD-score of 1.8 that could contain the pathogenic mutation, 1 of 70 cm on Xp22q13 and the other of 30 cm on Xq23q27.

Pedigree of family 1. X-linked pedigree of family 1 with 5 affected males over 3 generations. The mutation in KIF4A was shown to cosegregate in affected males (III:5, III:6, III:7 and IV:3, a DNA sample of II:5 was not available) and was not present in two unaffected males (III:3 and III:8). Females II:1, III:1 and IV:2 were carriers.
Massive parallel sequencing of the X-chromosome-specific exons revealed a likely pathogenic in-frame mutation, c.1489-8_1490delins10 (NM_012310.4), in KIF4A, which was predicted to disrupt the acceptor splice site of exon 15, leading to skipping of exon 15 and, consequently, an in-frame deletion in the protein (figure 2). KIF4A is located in the large Xp22q13 linkage interval. No rare deleterious mutations in known X-linked ID genes were identified. Sanger sequencing confirmed the presence of the KIF4A mutation, and cosegregation analysis showed that the mutation segregated with the phenotype in all affected males (a DNA sample of the uncle was not available). In addition, three females from three generations were shown to be carriers of this mutation and the mutation was absent in two unaffected males (figure 1). Analysis of the mutation at RNA level and sequencing of the cDNA of the PCR product showed that exon 15 is spliced out and skipped. (figure 3A,B). Further analysis at the protein level showed that KIF4A in the patient's cell line migrated with a lower molecular weight compared to the control (figure 3C). Importantly KIF4A expression level was also reduced with approximately 50% in the patient's cell line (figure 3D).
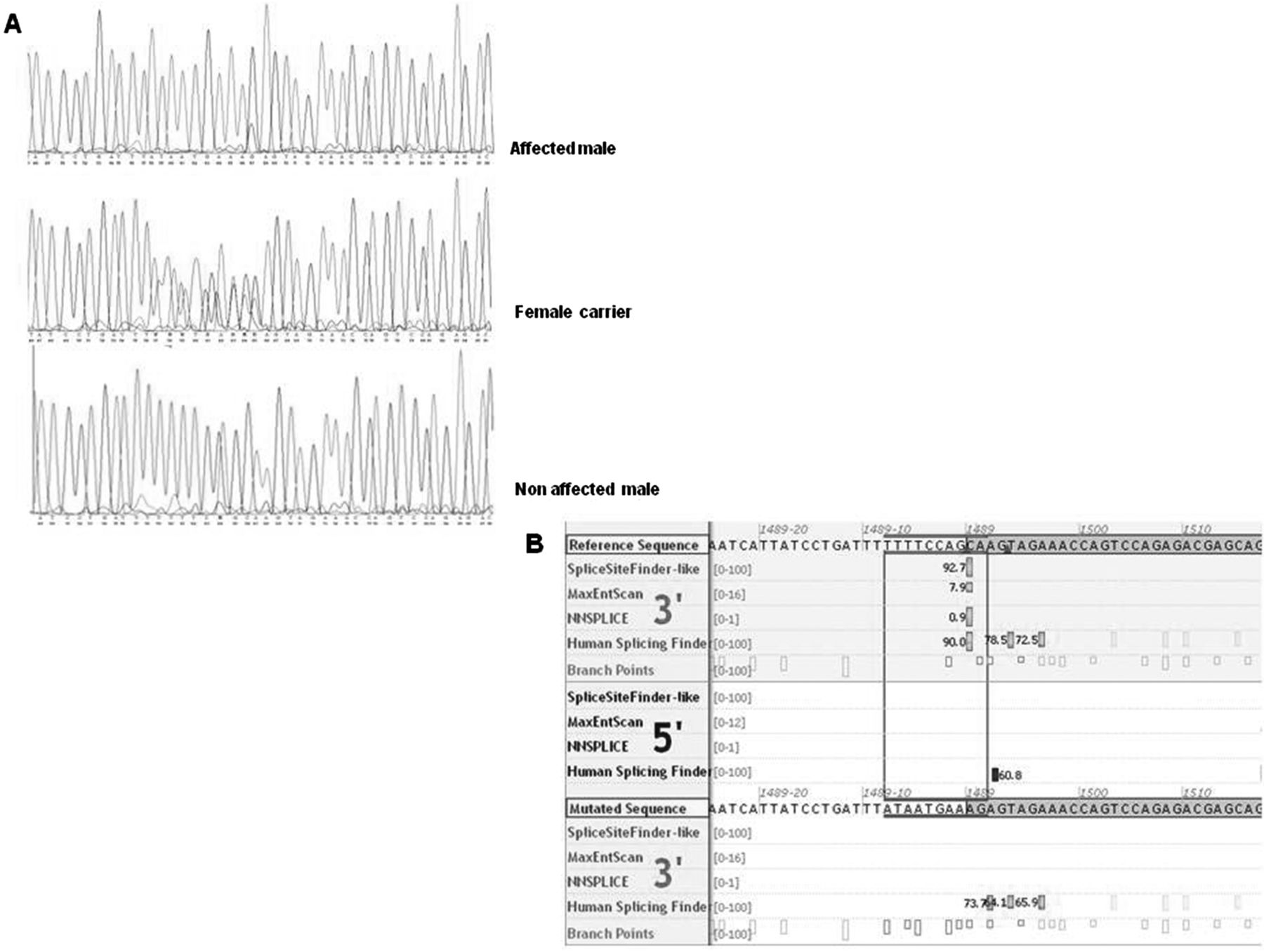
KIF4A sequences. (A) shows the sequences of affected males, carrier females and non-affected males. (B) shows the predicted splice acceptor sites (in green, upper part) of the reference sequence and the mutated sequence (in green, lower part), suggesting that the splice acceptor site of the mutated sequence is severely affected (Alamut Interactive Biosoftware).

RNA and protein expression studies in cell-lines of patients from family 1 with a mutation in KIF4A and control cell-lines, showing effect on splicing. (A, B) confirm that the KIF4 mutation indeed affects the acceptor splice site of exon 15, leading to a skip of exon 15 (186 bp in length), as predicted. RNA was isolated from a cell-line from an affected family member and a healthy control person. Primers for PCR analysis were designed at exon 13/14 (primers CTGCGGTGGAGCAAGAAGCC and AATTTGACCCTCCAGCTCCT, product size 340 bp) and exon 16/17 (CTGCGGTGGAGCAAGAAGCC and AATTTGACCCTCCAGCTCCT, product size 525 bp) bounderies and cDNA was synthesised. (A) Lane 1 shows the band of the patient (mt), which is 185 bp shorter in length as compared to the band in a healthy control (c), likely due to skipping of exon 15. (B) Sequencing of the PCR product of the patient sample showed that the sequence of exon 14 is directly followed by the sequence of exon 16 (upper part) and thus confirmed that exon 15 is skipped. The lower part of the figure shows the normal sequence including exon 15 in a control person. (C, D) confirm that the mutation leads to the production of a KIF4A protein with lower molecular weight in the patient (mt) compared to control (C), which corresponds to exon 15 skipping. C) Western blot showing the expression of KIF4A in control (lane 1 and 2) and patient cell line (lane 3 and 4). Gamma-tubulin was used as loading control. (D) Quantification of KIF4A expression level indicates that KIF4A is significantly reduced in the patients’ cell line. *p<0.01, t test.
Patient 2
Patient 2 was recently described in a large family based exome sequencing project performed by our group in a series of 100 patients with sporadic ID.26 Poirier et al19 also refer to this patient in their paper in which they report another family with a mutation in this gene. In short, patient 2 was born after an uncomplicated pregnancy and delivery with normal growth parameters. She had a severe developmental delay, did not acquire the ability to speak and could only walk with support since the age of 9–10 years. First seizures manifested at the age of 6 months. Her behaviour was characterised by severe automutilation. An MRI-scan of the brain at age 15 years showed signs of cortical malformation mainly affecting the frontal cortex, including a decreased number of gyri, coarse gyri and shallow sulci. (figure 4) These abnormalities were also seen to a lesser extent at the temporal cortex and insular cortex. During clinical evaluation at the age of 12 years she had a normal height (−1 SD), but a very dystrophic build and secondary microcephaly (head circumference: 49 cm (<−2.5 SD)). Additionally, she had small hands and feet. She had stereotypic hand movements and a slight hypertonia. There were no evident facial dysmorphisms. Because of the phenotypic similarities to Angelman-like and Rett-like syndromes, DNA analysis of MECP2 (MIM 300005), FOXG1 (MIM 164874), CDKL5 (MIM 300203), UBE3A (MIM 601623) and 15q11q13 methylation tests were performed, which were all normal. Subsequent 250k single nucleotide polymorphism (SNP) array analysis, DNA analysis of ARX, and a metabolic screen revealed no abnormalities either.

Cerebral MRI scan in patient 2, showing cortical malformation. (A, B) show signs of cortical malformation, mainly at the frontal cortex, including a reduction in the number of gyri, which have a coarse aspect, and shallow sulci (indicated by the arrows).
Family based exome sequencing revealed a highly conserved (PhyloP 6.1) de novo c.11465A>C mutation in KIF5C (NM 004522.1), predicting a p.(Glu237Lys) substitution. The Grantham score of 56 indicated a moderate physicochemical difference. The mutation affects the motor domain of the KIF5C protein, and may, in contrast to deletions previously reported for this gene,28 ,29 not lead to haploinsuffiency, but rather suggests a dominant negative effect resulting in abnormal heavy-chain dimerisation.
Effect of knockdown of Kif4a on mEPSC and mIPSC currents in rat primary hippocampal neurons
To find further supportive evidence that KIF4A is involved in the ID phenotype in family 1, we performed functional studies at the level of synaptic function in primary rat hippocampal neurons. This functional assay has previously been successful in showing that OPHN1 mediates mGluR-dependent long-term depression through interaction with the endocytic machinery.27 Interestingly, Kif4a knockdown using validated small interference (si)RNAs, resulted in a significant decrease in mEPSC amplitude, but an increase in mEPSC frequency compared to control siRNA (the BLOCK-iT fluorescent oligo) was used as transfection efficiency detector and a negative control. (Kolmogorov Smirnov (KS) test, p<0.001), suggesting that the development of excitatory synapses is hampered in neurons lacking Kif4a (figure 5A–C). Next, we measured mIPSCs, a response from quantal release of single GABAA vesicles. Down-regulation of Kif4a decreased mIPSC frequency but not amplitude, indicating that Kif4a affects presynaptic GABAA release and/or inhibitory synapse formation without affecting the amount GABAA receptors (GABAAR) at inhibitory synapses (figure 5A–C, KS-test, p<0.001). We did not observe any changes in the kinetics of mEPSCs or mIPSCs, indicating that the composition and/or function of the glutamate receptors and GABAAR subtype mediating the mEPSCs and mIPSCs, respectively, were unchanged (figure 5D,E).

Effect of KIF4A on mEPSC and miniature inhibitory postsynaptic currents (mIPSC). (A) Representative whole-cell current traces (mEPSC) of a control neuron or a neuron transfected with siRNAs against Kif4a at holding potential of −60 mV. Scale bars: 1.2 s and 20 pA. (B, C) Cumulative probability histograms of mEPSC amplitude and interevent intervals in control (n=39 neurons), Kif4a siRNA treated neurons (n=30 neurons). Differences between the conditions for the displayed mEPSC amplitude and interevent interval cumulative distributions are statistically significant (P<0.001, using Kolmogorov–Smirnov test). (D) Representative whole-cell current traces (mIPSC) of a control neuron or a neuron transfected with siRNAs against Kif4a at holding potential of −60 mV. Scale bars: 16 s and 25 pA. (E, F) Cumulative probability histograms of mIPSC amplitude and interevent intervals in control (n=7 neurons), Kif4a siRNA-treated neurons (n=10 neurons). Differences between control and Kif4a siRNA are statistically significant for the displayed interevent interval cumulative distributions but not for the mIPSC amplitude (P<0.001, using Kolmogorov–Smirnov test).
Effect of p. Glu237Lys mutation on KIF5C cellular localisation and function
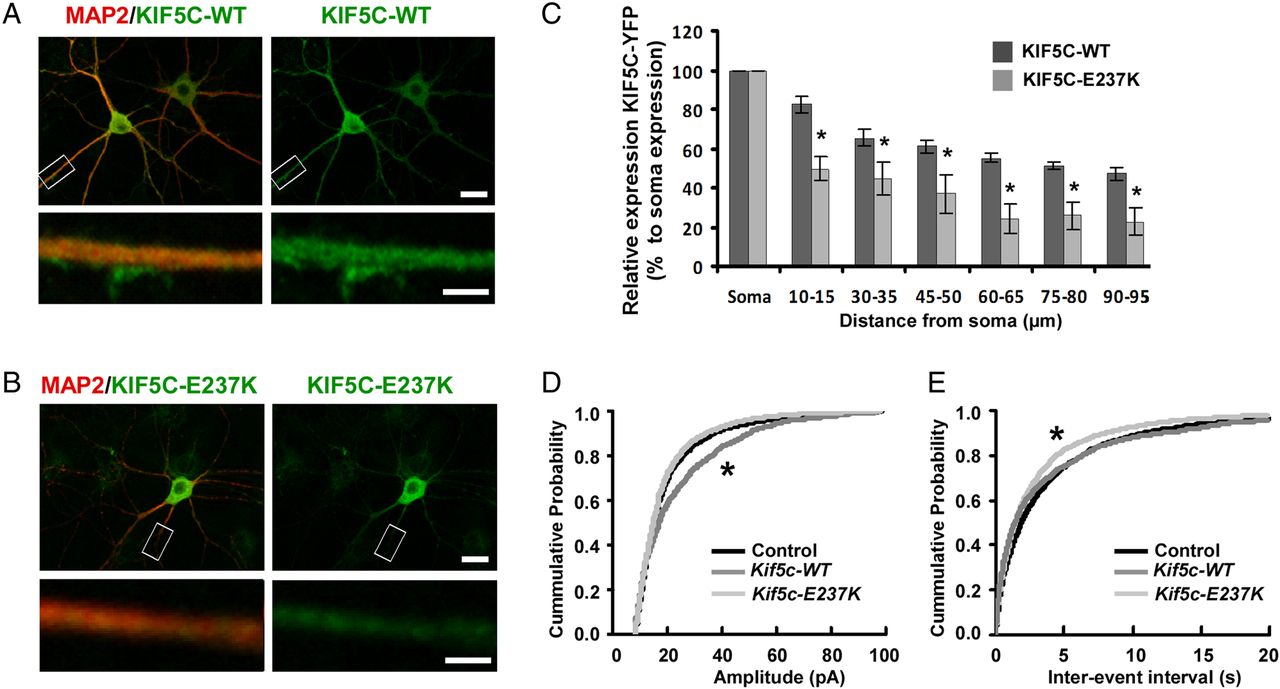
Since the mutation p.Glu237Lys in KIF5C is predicted to affect the motor domain, we first determined whether it affected KIF5c movement along dendrites. To this end, we transfected primary rat hippocampal neurons with Yellow Fluorescent Protein (YFP)-tagged wild-type KIF5C (KIF5C-WT) or mutant KIF5C (KIF5C-E237 K) construct. KIF5C-WT accumulated in distal regions of the dendrites (figure 6A,C), whereas KIF5C-E237 K showed a significant reduction in distal localisation and a relative increased accumulation throughout the cell body (figure 6B,C, t test p<0.05). Next, to determine the functional consequence of the missense mutation, we compared the effect of overexpressing KIF5C-WT versus mutant KIF5C-E237 K and control (YFP only) on mEPSCs in hippocampal neurons. Overexpressing KIF5C-WT resulted in an increase in mEPSC frequency compared to control transfected neurons (figure 6D,E, KS-test, p<0.001). However, overexpressing KIF5C-E237 K failed to increase mEPSC frequency, but by contrast, led to a significant decrease in mEPSC amplitude. Together, these data indicate that the missense mutation found in KIF5C leads to a non-functional motor domain.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Impact of p.Glu237Lys on KIF5C localisation and function. (A, B) Representative images showing the subcellular localisation of Yellow Fluorescent Protein (YFP)-tagged KIF5C-WT and KIF5C-E237 K. YFP-tagged constructs of KIF5C were expressed in cultured hippocampal neurons and visualised by immunofluorescence staining with an anti-GFP antibody. KIF5C-WT accumulated in the distal regions of the dendrites, by contrast with KIF1C-E237 K. The neuronal cell bodies and dendrites were stained with anti-MAP2 antibody. Scale bars upper image: 20 μm, lower image: 4 μm. (C) Quantification of dendritic expression of YFP-tagged KIF5C-WT and KIF5C-E237 K. Dendritic signal of KIF5C-WT and KIF5C-E237 K was normalised to its signal in the soma (p<0.05, using t test, n=6 neurons). (D, E) Cumulative probability histograms of mEPSC amplitude and interevent intervals in control (n=30 neurons), KIF5C-WT expressing neurons (n=13 neurons) and KIF5C-E237 K expressing neurons (n=27 neurons). Differences between the conditions for the displayed mEPSC amplitude and interevent interval cumulative distributions are statistically significant (p<0.001, using Kolmogorov–Smirnov test).
Discussion
The role of KIF genes in the origin of ID has so far only received limited attention, although a growing body of work suggests that many ID genes impinge on synaptic function and, as such, contribute to the pathophysiology of the disorder.30 In particular, the balance between excitatory and inhibitory synaptic input seems to be disturbed in common neurodevelopmental disorders including ID, autism and schizophrenia.
Our data show that both KIF4A and KIF5C are important proteins that regulate synapse development. In particular, KIF4A is a critical protein in controlling the tight balance between excitatory and inhibitory inputs during development, since knocking down Kif4a resulted in a decrease in mIPSCs, but an increase in mEPSCs, albeit with a lower amplitude. A disturbed balance between excitatory and inhibitory drive could, therefore, explain the presence of epilepsy in the patients. Moreover, we showed that the reported missense mutation in Kif5c affects its protein function at excitatory synapses.
A synaptic function for KIF4A has so far not been explored, whereas KIF5 was recently reported to be involved in regulating both glutamatergic and GABAergic synaptic transmission in striatal and cortical neurons.31–33 The specific contribution of KIF5C hereto has, however, not been determined.
KIF4A belongs to the class of the N5-kinesins and is highly expressed in differentiated young neurons. It consists of a N-terminal motor domain, a central stalk domain and a C-terminal C-domain.34 Previous studies showed that KIF4A has various functions and plays a role in chromosome condensation and midzone spindle pole formation essential for cytokinesis and chromosome segregation during mitosis, as well as in regulation of activity-dependent neuronal survival and apoptosis, thereby regulating the number of neurons during murine brain development.9 ,35–37 After dissociation from the enzymatic protein poly (ADP-ribose) polymerase 1 (PARP1), which prevents apoptosis, KIF4A moves into the neurites. Here, it possibly transports cargoes essential for neuronal development, such as the adhesion molecule L1 which is required for axon formation and synapse development.9 ,38 Although KIF4A was shown by several studies to be involved in spindle pole organisation, most studies concluded that KIF4A deficient cells show a normal mitotic division. Aneuploidy analysis in cultured blood lymphocytes from one of the patients of family 1 showed no increased level of aneuploid cells, which is in agreement with the observation in most other studies that mitosis is not significantly disturbed.9 ,35 ,36
KIF5C, together with KIF5A and KIF5B, belongs to the class of N-1 kinesins, and is specifically expressed in nervous tissue.1 ,13 ,39 Mice with a homozygous knockout of Kif5c are viable and do not show gross abnormalities in the nervous system, except from a smaller brain size and relative loss of motor neurons to sensory neurons. This mild phenotypic presentation is explained by a high similarity between the three KIF5 genes and functional redundancy.13
The missense mutation in the family reported by Poirier et al19 is associated with a severe phenotype including severe MCD and microcephaly, which shows remarkable overlap with the phenotype in the present patient 2, who also presented with microcephaly and has signs of cortical malformation on her brain MRI as well. Interestingly, Poirier et al showed that expression of the missense variant that they found in the family with MCD (p.Glu237Val) in Escheria Coli, results in a complete inability of the protein to hydrolyse adenosin triphosphate in a microtubule-dependent assay. Of note, this missense variant is located at the same position as the here-reported variant, and affects the microtubule-interacting domain. Additionally, they found an altered distribution of the protein. These data are in agreement with our results that show that the missense mutation in KIF5C leads to a non-functional motor domain, a significant reduction in distal localisation of the protein and a relative increased accumulation throughout the cell body.
In summary, these data support the involvement of members of the kinesin superfamily, KIF4A and KIF5C, in ID phenotypes. Identification of other patients with mutations in these genes should further confirm the role of KIF4A and KIF5C in ID phenotypes and may give further insights in the full clinical spectrum. This spectrum likely includes neuronal migration defects as was supported by the aberrant cortical formation in patient 2 and the previously reported family. At the functional level, we found that both KIF4A and KIF5C may contribute to the pathophysiology of ID by altering the balance between excitatory and inhibitory synaptic efficacy leading to changed neuronal excitability. As such, these genes can be added to the growing list of ID genes that influence synaptic functioning.30 Together with recently reported mutations in similar and other proteins involved in the motor protein complex of the microtubule transport pathway, including DYNC1H1, KIF2A and TUBG1,19 ,40 our findings indicate that this pathway may be of great importance to ID, and suggest that other genes encoding proteins in this pathway may well be potential candidates for ID phenotypes.
Acknowledgments
We thank the participating patients and their families. We also would like to thank Michél AAP Willemsen and Jolanda H Schieving, from the Department of Pediatric Neurology of the Radboud University Medical Centre, Nijmegen, The Netherlands, for their expert evaluation of the brain MRI in patient 2. We thank Dr Anne Stephenson for providing YFP-KIF5C constructs.
References
Footnotes
-
MHW, WB, NNK and TK contributed equally.
-
Contributors MHW, NNK and TK designed the study and wrote the manuscript. WB and NNK performed the synaptic investigations and western blots. WMW-L performed the RNA expression studies of KIF4 and confirmed the mutation in KIF4A by Sanger Sequencing. SAH, MB, HH and VK designed and performed the X-exome studies that were performed in family 1. LELMV was involved in the trio-sequencing project in which the mutation in KIF5C in patient 2 was detected.
-
Funding This work was supported by grants from the Consortium ‘Stronger on your own feet’ to TK and MHW, The Netherlands Organisation for Health Research and Development (ZonMw grants 916-86-016 to LELMV, and 907-00-365 to TK), the GENCODYS, an EU FP7 large-scale integrating project grant (241995 to HvB and TK) and the MERE-GLU, an EU FP7 Marie Curie Re-integration Grant (PEOPLE-2010-RG, 277091 to NNK).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Local medical ethical committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
WEB RESOURCES
-
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org