Article Text

Abstract
Background Despite abundant evidence for pathogenicity of large copy number variants (CNVs) in neurodevelopmental disorders (NDDs), the individual significance of genome-wide rare CNVs <500 kb has not been well elucidated in a clinical context.
Methods By high-resolution chromosomal microarray analysis, we investigated the clinical significance of all rare non-polymorphic exonic CNVs sizing 1–500 kb in a cohort of 714 patients with undiagnosed NDDs.
Results We detected 96 rare CNVs <500 kb affecting coding regions, of which 58 (60.4%) were confirmed. 6 of 14 confirmed de novo, one of two homozygous and four heterozygous inherited CNVs affected the known microdeletion regions 17q21.31, 16p11.2 and 2p21 or OMIM morbid genes (CASK, CREBBP, PAFAH1B1, SATB2; AUTS2, NRXN3, GRM8). Two further de novo CNVs affecting single genes (MED13L, CTNND2) were instrumental in delineating novel recurrent conditions. For the first time, we here report exonic deletions of CTNND2 causing low normal IQ with learning difficulties with or without autism spectrum disorder. Additionally, we discovered a homozygous out-of-frame deletion of ACOT7 associated with features comparable to the published mouse model. In total, 24.1% of the confirmed small CNVs were categorised as pathogenic or likely pathogenic (median size 130 kb), 17.2% as likely benign, 3.4% represented incidental findings and 55.2% remained unclear.
Conclusions These results verify the diagnostic relevance of genome-wide rare CNVs <500 kb, which were found pathogenic in ∼2% (14/714) of cases (1.1% de novo, 0.3% homozygous, 0.6% inherited) and highlight their inherent potential for discovery of new conditions.
- Genome-Wide
- Copy-Number
- Developmental
- Diagnostics
- Clinical Genetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Neurodevelopmental disorders (NDDs) are a group of conditions characterised by cognitive, neurological and/or psychiatric manifestations occurring during the development of the nervous system.1 The various clinical entities such as intellectual disability (ID), speech and language delay, autism, neuromotor dysfunction or epilepsy show considerable comorbidity and may be associated with a variety of non-neurological features within complex syndromes.2 ,3 Due to the extensive aetiological heterogeneity of NDDs, the majority of patients remain without aetiological diagnosis, which hampers disease management, genetic counselling and in-depth studies of the underlying molecular mechanisms. With the advent of new genomic technologies, however, diagnostic yield is steadily improving and a rapidly growing number of novel, aetiologically defined disorders are delineated.
Genome-wide chromosomal microarray analysis (CMA) for detection of copy number variants (CNVs) is currently used as a first-tier diagnostic approach in patients with idiopathic NDDs. The diagnostic yield of clinically significant CNVs varies between 5% and 20%, depending on the clinical preselection and resolution of the array.4 Despite their obvious higher sensitivity, the widespread use of high-resolution arrays, however, is hampered by their inherent burden of detecting polymorphic or unclear variants. Indeed, tiling array studies have revealed a huge diversity of CNVs in the general population with an overall median length of about 2.9 kb and 95% being less than 100 kb.5 Furthermore, CNVs larger than 500 kb were shown to occur only in about 10% of control individuals, while patients with NDDs harbour an additional burden of more than 13.5% for such CNVs.6 Accordingly, a 2010 consensus statement on diagnostic chromosomal microarray testing recommends a resolution of ≥400 kb throughout the genome as a balance of analytical and clinical sensitivity.4 Therefore, the individual significance of rare small CNVs has not been well elucidated in a clinical context, but is of rising interest given the recent progress in detection of small CNVs from whole-exome sequencing (WES) data.7–9
Therefore, in this study, we investigated the diagnostic relevance and inherent potential for gene discovery of rare CNVs sizing 1–500 kb in a cohort of 714 patients with isolated or syndromic NDDs.
Methods
Excluding patients with large-scale chromosomal aberrations, CNVs >10 Mb, or clinically recognised recurrent microdeletion syndromes, we investigated 714 patients with NDDs with or without further congenital anomalies by genome-wide high-resolution CMA. The vast majority of patients were of European origin. Among them, 63 patients (8.8%) had obvious pathogenic CNVs >500 kb with a median size of 3.8 Mb.
We investigated CNVs sizing 1–500 kb for their overlap with annotated exons as well as with in-house and public control databases. CNVs affecting exonic regions that were not observed in our in-house controls or only reported once in public databases were tested by multiplex ligation-dependent probe amplification (MLPA) or fluorescence in situ hybridization (FISH) if not already confirmed by inheritance pattern from trio microarray analysis. Confirmed CNVs were individually assessed regarding literature evidence for pathogenicity, overlapping CNVs in the DECIPHER database,10 function and expression profiles of the affected gene(s) and inheritance pattern. Selected candidate genes within inherited rare CNVs were further studied by Sanger sequencing for biallelic mutations. Five patients were further investigated for non-allelic hits by WES. Four of these patients were selected for WES because they had de novo CNVs affecting good candidate genes but lacking overlapping cases at the time of analysis, and one patient was exome sequenced because an inherited variant was present in three affected siblings.
Microarray and confirmatory studies
DNA, extracted from peripheral blood, was analysed with Affymetrix Genome-Wide Human SNP Array 6.0 (1.8 million markers; 79 patients), Affymetrix Cytogenetics 2.7 (2.7 million markers; 423 patients) and CytoScan HD (2.6 million markers; 212 patients) (Affymetrix Inc., Santa Clara, California, USA). The average intermarker spacing was 1.6 kb for the 6.0 array and about 1.1 kb for the two other arrays. CNVs were called if they encompassed at least five consecutive markers resulting in a maximum resolution of about 2 kb. The data set of each patient's sample was evaluated with Affymetrix Chromosome Analysis Suite (ChAS V.1.0.1) in comparison with 670 controls in the 6.0 array, 820 controls in the 2.7 array and 1038 controls in the CytoScan array. Controls consisted of European and American healthy individuals. Categorisation of CNVs by the Affymetrix's ChAS software among others includes confidence values for the 2.7 and CytoScan arrays. Confidence is determined on a marker by marker basis by evaluating the concordance of the log2ratio at each marker with the copy number state assigned by the hidden Markov model (HMM). The average confidence score of markers in gain and loss segments determines the confidence score of that segment.
Readily available kits or customised MLPA was performed using synthetic probes for selected exons and the SALSA MLPA kit P300 Human DNA reference-2 (MRC-Holland, Amsterdam, The Netherlands). The MLPA module of the Sequence Pilot 3.5.2 Build 508 software (JSI medical systems GmbH, Kippenheim, Germany) was used to retrieve relative peak intensities by normalisation to the reference probe set. Normalised peak levels were set in relation to at least three healthy control individuals. FISH analyses were performed using locus-specific commercial probes according to standard protocols on metaphase preparations from peripheral blood.
SATB2 protein modelling
The protein was modelled with Modeller 9.9,11 based on the crystal structure of the homologous SATB1 tetramer 12 that exhibits 78% sequence identity.
Exome sequencing and mutation analysis
WES on genomic DNA of selected patients was performed as described before with minor modifications.13 ,14 All exons and flanking intronic nucleotides of candidate genes from CMA or candidate nucleotide variants from WES were analysed after PCR amplification from patient's DNA by Sanger sequencing using an ABI Genetic Analyzer 3730 (Applied Biosystems, Foster City, California, USA).
Expression studies of ACOT7
Expression levels were investigated in cDNA panels from fetal and adult human tissues using customised SYBR green qPCR for exons 1 and 2 of ACOT7 (specific for isoform ENST00000377855). Relative expression levels normalised to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were set into relation to the mean expression value of this isoform in fetal brain.
Statistical analysis
Statistical differences for size, number of markers/size and confidence value of CNVs were analysed using Mann-Whitney U-testing and independent-samples t testing. p Values less than 0.05 were considered statistically significant.
Results
Among the 714 array results, 96 aberrations below 500 kb fulfilled the abovementioned criteria and were further evaluated (size range 2–492 kb, median 72 kb). In total, 58 out of 96 (60.4%) of these selected CNVs were confirmed by secondary testing, while 38 of 96 (39.6%) were not confirmed and thus considered false positive (see online supplementary table S1). False positive aberrations were significantly smaller in size (3–181 kb, median 19 kb, mean 45.3 kb) than true CNVs (2–492 kb, median 131 kb, mean 164.7 kb) (p<0.0001; figure 1A). There was also a significant difference between the two groups regarding their confidence values (mean of 88.9% vs 91.9%, p<0.0001; figure 1B) and marker count (median of 20 (8–142) vs 96 (6–1040), mean of 39.9 vs 163.8, p<0.0001; figure 1C), while no significant difference was observed for the marker count per kb within the CNV (1.5±1.14 vs 1.1±0.67, p=0.1; figure 1D). Since the 1:2 copy number reduction in deletions is more easily detectable than the 3:2 copy number gain in duplications, sensitivity and specificity is different for deletions and duplications. Duplications sizing at least 183 kb or encompassing at least 168 markers were all true positives, while deletions were reliable if they sized at least 113 kb or encompassed at least 52 markers.
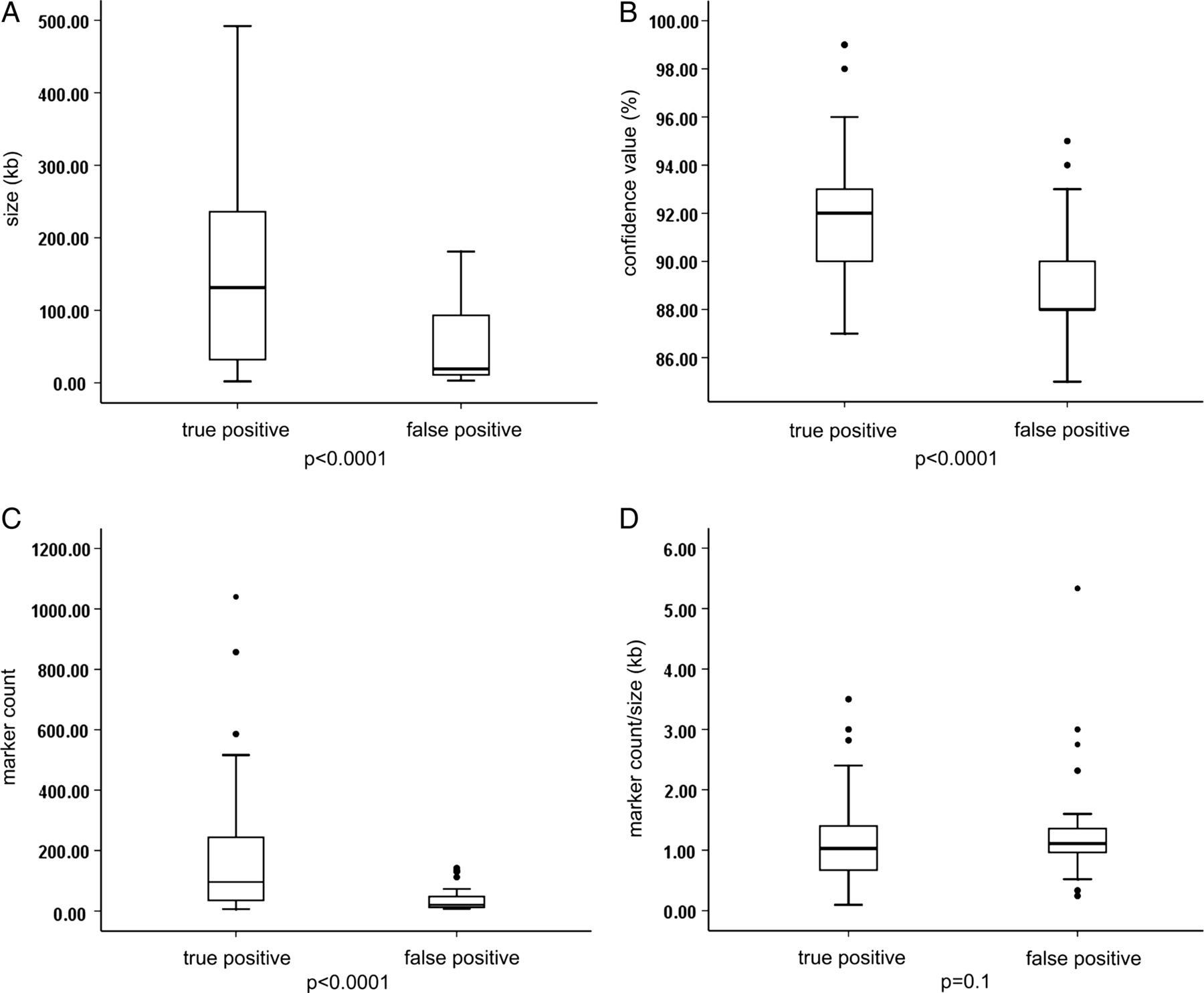
Comparison between true versus false positive status of small copy number variants (CNVs) detected by chromosomal microarray analysis (CMA) and their size, confidence value, marker count and marker count per kb. (A) False positive CNVs were significantly smaller in size (3–181 kb, median 19 kb, mean 45.3 kb) than true CNVs (2–492 kb, median 131 kb, mean 164.7 kb) (p<0.0001). (B) There was also a significant difference between the two groups regarding their confidence values (mean of 88.9% vs 91.9%, p<0.0001) and (C) marker count (median of 20 vs 96, mean of 39.9 vs 163.8, p<0.0001). (D) No significant difference was observed for the marker count per kb within the CNV (1.5±1.14 vs 1.1±0.67, p=0.1).
Among 58 confirmed CNVs, 14 (24.1%) were de novo (table 1), 2 (3.4%) were homozygous and 39 (67.2%) were heterozygous and inherited (19 from mothers and 20 from fathers). While for 12 of 14 de novo CNVs both parents were available for testing, in another two CNVs de novo origin was assumed based on their well-established causal involvement in severe and fully penetrant phenotypes (one exonic deletion within the PAFAH1B1 gene causing lissencephaly type 1 and the recurrent 473 kb microdeletion in 17q21.31) (table 1). For two further CNVs (3.4%), patterns of inheritance could not be completely tested because the fathers were not available, but due to familial recurrence they were considered likely inherited (see online supplementary table S2). One incidental finding (DMD deletion) was not tested in the mother.
Clinical and genetic features of patients with candidate de novo CNVs <500 kb sorted by descending size
De novo CNVs
Six de novo or likely de novo CNVs were clearly pathogenic affecting the recurrent microdeletion region in 17q21.31 (detected in two cases), well-characterised OMIM morbid genes (CASK, CREBBP, PAFAH1B1) or the recently described SATB2 locus (table 1). For two further de novo CNVs affecting single genes (MED13L, CTNND2), similar cases were identified in the DECIPHER database (http://decipher.sanger.ac.uk/). The 17 kb out-of-frame de novo deletion encompassing exon 2 of MED13L (MIM *608771) in patient 56366, together with overlapping cases, were instrumental to define a recognisable haploinsufficiency syndrome that we reported and discussed in detail elsewhere.13 The novel condition caused by CTNND2 haploinsufficiency is described for the first time below. We also report a special tooth phenotype found in our patient with SATB2 defect. Additionally, we discuss a de novo variant limited to the DNM3 gene, which is the candidate critical gene in 1q24-q25 deletions, as well as two de novo CNVs classified as likely benign after identification of pathogenic mutations by WES.
Novel CTNND2-related phenotype defined by patient 62563 with 113 kb deletion and overlapping cases
CTNND2 (MIM *604275) encodes δ-catenin, which functions as a regulator of neuronal migration15 and maintenance of dendrites and dendritic spines in mature cortex.16 It was mapped to the cri-du-chat syndrome critical region in chromosome 5p15.2 and was considered responsible for severe ID in typical cri-du-chat syndrome patients with terminal 5p deletions.17 However, extended deletion mapping indicated that interstitial deletions restricted to the ID critical region 2 (MRII) including the CTNND2 locus produce a milder level of intellectual impairment.18 CNVs encompassing CTNND2 have been implicated in autism (one deletion, de novo),19 cerebral palsy (one duplication including the first exon of CTNND2, maternally inherited)20 and schizophrenia (one duplication affecting seven genes including CTNND2).21
CMA in our patient revealed a 113 kb de novo out-of-frame deletion encompassing exons 4–7 of CTNND2. Sanger sequencing in the patient did not reveal an additional pathogenic point mutation of the gene. The girl was born spontaneously at term with normal measurements and no complication to highly educated unrelated parents. She had no remarkable health issues and developmental milestones and growth parameters were within normal limits (table 2). Physical examination revealed deep set eyes, prominent cheeks, narrow eyebrows, short inner canthal distance (ICD 2.7 cm, 2nd centile), low-set, slightly backwards rotated ears, and a bulbous nose with prominent columella. She had mild clinodactyly of the fifth finger, which was also present in the healthy brother. She was referred to formal developmental testing at age 8 years because of behavioural issues and was diagnosed with borderline ID (WISC-IV, full scale IQ 77). She showed a dissociated cognitive profile with better language (vocabulary, comprehension, reading) than non-verbal functions (visual perception, abstract reasoning). She also showed short attention span, poor executive functioning and impaired working memory. The cognitive profile remained stable at follow-up. Although immature social-emotional behaviour was described, formal signs of autism were not present.
Summary of the patients with deletions affecting CTNND2
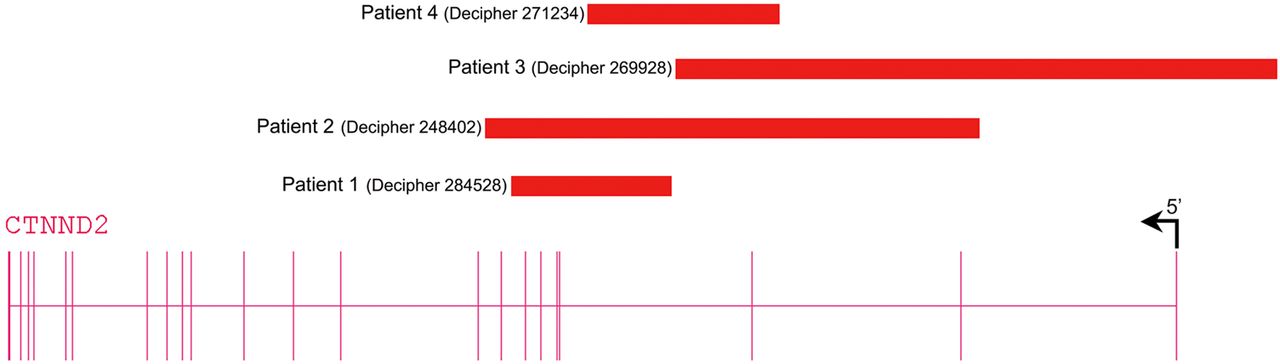
We found three further patients with exonic deletions limited to CTNND2 via the DECIPHER database (table 2, figure 2). These three deletions were inherited and for two transmitting parents low normal IQ was recognised. Because exonic CTNND2 deletions are not reported in the normal population databases and since these patients as well as two of the three transmitting parents share borderline low IQ or mild ID with or without autistic behavioural problems, we assume that CTNND2 haploinsufficiency is causing the neurodevelopmental features in these patients. Given the mild phenotype of the patients, transmission by seemingly normal parents may be explained by clinical variability or lack of formal cognitive testing. The progressive neurological signs in patient 4 (DECIPHER 271234), however, may be caused by an additional unidentified disorder.

Schematic representation of CTNND2 deletions detected in patients. Patient 1 (DECIPHER #284528) with a de novo deletion of exons 4–7, patient 2 (DECIPHER #248402) with a paternally inherited deletion encompassing exons 2–8, patient 3 (DECIPHER #269928) with a maternally inherited deletion of exons 1–3 and 5'UTR and patient 4 (DECIPHER #271234) with a maternally inherited deletion of exon 3.
SATB2 intragenic 32 kb duplication in patient 70886 with ID and double row of upper incisors
SATB2 (special AT-rich sequence binding protein 2; MIM *608148) is a DNA-binding protein that regulates gene expression and corticocortical connections in the developing cerebral cortex and craniofacial patterning.22 ,23 De novo interrupting translocations, microdeletions and mutations of SATB2 have been described in patients with ID, behavioural problems, seizures and craniofacial anomalies with or without cleft palate.14 ,24–28 The duplication detected by CMA in our patient affects exon 4 (c.170-?_346+?; p.Gly57_Gln115dup) (ENST00000457245) and is predicted by homology modelling to disturb protein tetramerisation, which plays an important role for long-range chromatin organisation and coordination in gene regulation (figure 3A).12

Structure of the Satb2 tetramerisation domain and upper incisors in the patient with intragenic SATB2 duplication. (A) The four subunits of the tetramer are shown in different colours and those parts, which are duplicated in the mutant, are shown in space-filled presentation. This duplication will affect the interfaces between the dimers that form the tetramer (black arrows). Thus, the intragenic duplication is expected to hamper formation of the tetramer, which was suggested to play an important role for long-range chromatin organisation and coordination in gene regulation.12 (B) Double row of upper incisors in the patient with 32 kb pathogenic duplication within SATB2 at age 3 3/12 years.
This patient was a 4-year-old boy born at term with normal measurements (51 cm; 3330 g) to healthy unrelated parents. After birth hypotonia, feeding difficulties and a cleft soft palate were noted and at age 1 year a double row of upper incisors became evident (figure 3B). At age 3 3/12 years, he was remarkably hypotonic and had borderline microcephaly (OFC 49 cm, 3rd centile) with normal height and weight. The face was long, flat and hypotonic with hypersalivation, an impression of mild hypertelorism, down-slanting palpebral fissures, mild ptosis, flat nasal bridge, anteverted nares, long flat philtrum and mild micrognathia. He had large ears, low posterior hair line, mild pectus excavatum, mildly wide spaced mammillae, bilateral 5th finger camptodactyly, mild cutaneous syndactyly of toes 2 and 3, and a 1×1.5 cm depigmented spot on the leg. The facial features of this patient resembled patient 1 with a SATB2 deletion published by Rosenfeld et al.26 Psychomotor development was mildly delayed with walking age 18 months and fine motor problems, but expressive speech development was remarkably delayed with few single words, hyperactivity and stereotypic movements at the age of 4 years.
Although a doubled row of upper incisors has not been previously described in SATB2 defects, some patients had tooth abnormalities such as missing teeth, abnormally shaped and crowded teeth, malocclusion and diastema. Moreover, in E17.5 Satb2−/− mouse embryos the incisor teeth, which express high levels of SATB2 in the wild type, were missing, while the molars, which do not express SATB2 in the wild type, were unaffected.23
DNM3 intragenic 372 kb deletion of uncertain significance in patient 59248 with epileptic encephalopathy
DNM3 (MIM *611445) functions in endocytosis of presynaptic vesicles after release of neurotransmitter and postsynaptic receptors,29 and there is evidence for its interaction with mGluR5 and Homer and its role in dendritic spine morphogenesis.30 DNM3 was considered the critical gene for the neurodevelopmental features in patients with larger deletions of 1q24q25, which in addition to severe cognitive disability show a recognisable phenotype including prenatal-onset microcephaly, growth deficiency, small hands and feet with distinctive brachydactyly and distinctive facial features.31
CMA showed a 372 kb de novo deletion within the gene DNM3, encompassing exons 2–15 in our patient. This girl was referred for severe hypotonia since birth, profound developmental delay, refractory seizures, oedematous hands and feet with tapering fingers and facial dysmorphism. Because of severe epileptic encephalopathy and oedema but absence of typical features such as cerebellar and optic atrophy, a clinical diagnosis of progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO)-like syndrome was proposed. However, CNV analysis and Sanger sequencing of DNM3 in eight similar patients with PEHO or PEHO-like syndrome did not reveal any pathogenic finding. This deletion remains of uncertain significance because seizures are only reported in a minority of patients with larger 1q24-q25 deletions and because of the mild phenotype consisting of attention deficit hyperactivity disorder (ADHD) and autism in DECIPHER patient 288412 with a 400 kb deletion limited to DNM3.
WES of our patient and both healthy parents revealed a heterozygous de novo missense mutation in ADAM7 (c.190A>G; p.K64E; chromosome 8g.24304732A>G). Since no germline mutation has been reported so far for ADAM7, the relevance of this finding remains also unclear.
Likely benign 19 kb de novo deletion affecting TH1L (NELFCD) and CTSZ in patient 45333
Both genes (MIM *605297 and *603169) are widely expressed in fetal and adult tissues32 and TH1L interacts with A-Raf kinase, an important intermediate of the growth factor Ras-MAP kinase pathway.33 CMA revealed a 19 kb de novo deletion partially affecting TH1L and CTSZ. This boy had macrocephaly, severe ID, hypotonia, haemangioma of the upper lip, bilateral postaxial foot polydactyly and obesity. There was no strong evidence for the pathogenicity of this aberration, and larger overlapping deletions reported in the DECIPHER database (eg, numbers 250209 and 250318) had a divergent phenotype of short stature and microcephaly. Eventually WES revealed the recently reported PIK3CA, c.2740G>A, p.G914R mutation in mosaic form causing the megalencephaly capillary malformation syndrome,34 which fully explains the patient's phenotype. The mutation was confirmed by Sanger sequencing in blood (∼7%) and saliva (∼16%) and occurred de novo.
Likely benign 122 kb de novo duplication affecting TAOK3 and PEBP1 in patient 62848
TAOK3 encodes the serine/threonine-protein kinase TAO3 that acts as a regulator of the p38/MAPK14 stress-activated MAPK cascade involved in the G2/M transition DNA damage checkpoint.35 PEBP protein is an inhibitor of the Raf/MEK/MAP kinase signalling cascade and functions in the regulation of cell cycle.36 Therefore, both genes have critical roles in the regulation of cell cycle and can be dosage sensitive. CMA showed a de novo 122 kb duplication within TAOK3 and PEBP1 (MIM *604591) in a girl with microcephaly, mild developmental delay and hyperactivity. So far, no polymorphic variants overlapping with this duplication have been reported and a smaller duplication limited to TAOK3 was reported in DECIPHER (#250362) in a patient with microcephaly and developmental delay, but was reported as inherited from a healthy parent. Eventually WES revealed the SHANK2 (ENST00000338508) de novo c.2669_2670insC mutation, which causes a frameshift introducing a premature stop codon (p.P891Sfs*32), and explains the patient's phenotype. However, we cannot exclude a multiple hit aetiology in this patient.
Homozygous CNVs
We found two rare homozygous deletions with heterozygous healthy parents (see online supplementary table S2). One, an 83 kb homozygous deletion on 2p21 in two siblings with hypotonia-cystinuria syndrome without cystinuria, further refined the genotype–phenotype correlation in this known ID region and was described and discussed in detail elsewhere.37
The second was a 7 kb homozygous frameshift deletion encompassing the first exon of ACOT7 (isoform ENST00000377855) in a patient with ID, epilepsy and abnormal behaviour. So far no human disorder has been described for any of the ACOT proteins. This boy was born at 40+9 weeks of gestation with normal measurements (3780 g, 53 cm). At age 16 months, he developed a generalised mixed myoclonic-tonic absence seizure disorder. Anticonvulsive treatment was discontinued at age 8 years without recurrence of seizures. Since age 15 years, episodes with ravenousness, extreme fatigue and fluctuating alertness were noted and led to cardiologic evaluation without abnormal findings. He had normal body measurements, micrognathia and mild ID with an IQ of 55–65, hyperactivity and abnormal behaviour and spoke in simple sentences but could not take care of himself.
ACOT7 (MIM *602587), formerly known as brain acyl-CoA hydrolase (BACH), encodes acyl-CoA thioesterase 7 and is involved in fatty acid metabolism with other ACOTs.38 ACOT7 encodes distinct isoforms with tissue-specific expression and subcellular locations and is strongly expressed in human brain cells such as pyramidal cells in the cerebral cortex, as well as in testes and some other tissues.39 ,40 Although lowered levels of ACOT7 in patients with suspected mitochondrial fatty acid oxidation disorders have been shown41 and a derangement of the ACOT7 protein has been detected in the hippocampus of patients with mesial temporal lobe epilepsy,42 so far, no mutation or CNV within ACOT7 was linked to any particular disorder. Recent studies of ACOT7 conditional central nervous knockout mice (KO) showed that ACOT7 counter-regulate fatty acid metabolism in neurons and protects against neural lipotoxicity. Interestingly, the KO mice exhibited behavioural hyperexcitability after fasting when circulating free fatty acids from lipolysis are elevated,43 which resembles the episodes of ravenousness and fatigue observed in our patient.
In our patient, the homozygous frameshift deletion within the alternatively spliced first exon of isoform ENST00000377855 most likely results in the depletion of its transcription. In other isoforms, however, it is intronic or in the 5'UTR and the possible effect on transcription or splicing remains uncertain. To further elucidate the involvement of this ACOT7 isoform in the patient's phenotype, we investigated its expression in cDNA panels from fetal and adult human tissues and found the highest levels in adult pancreas, testis, brain, lung, prostate and colon (see online supplementary figure S1). Since we found no expression in control lymphoblasts, we were not able to perform expression studies in the patient. Given the similarity to the KO mice phenotype and the segregation of the deletion in the family with heterozygosity in both healthy parents and the healthy brother and absence of the deletion in the healthy sister, it is likely that the homozygous deletion is pathogenic. Since the same deletion has been observed in the heterozygous state in 1 out of 1038 worldwide Affymetrix controls (∼0.1%), in 1 out of 451 controls by Conrad et al5 (∼0.2%) and in 13 out of 1151 of 1000 Genomes Consortium controls (∼1.1%),44 the homozygous disease frequency would be ∼0.000025–0.003%, which is in line with a very rare disorder (1:33 000–4 000 000).
Inherited heterozygous CNVs
Four inherited CNVs were considered as pathogenic or likely pathogenic affecting genes with reported pathogenic rare de novo/inherited deletions (AUTS2, NRXN3), deletions of a gene observed only in ADHD patients but not in controls (GRM8) or the recurrent microdeletion/duplication region in 16p11.2. These CNVs are described and discussed in the supplementary information. Six of the familial CNVs were found to have some evidence for potential pathogenicity but remained with uncertain significance (VOUS) including one duplication in 20p13 and five deletions affecting STPG2 (C4orf37), SUCLG2, PARK2, NDUFV3 and WDR4, and TPK1, respectively.
Eight CNVs were considered likely benign because of the identification of independent pathogenic mutations fully explaining the phenotype or observation of similar CNVs in new control data (see online supplementary table S2). For the rest of inherited CNVs (22), there was no evidence in favour or against pathogenicity (see online supplementary table S1). Selected genes of 10 familial CNVs suspected for recessive pathogenicity were sequenced for a second hit in the trans allele, which was negative for any pathogenic finding (see online supplementary tables S2 and S3).
Incidental findings related to NDDs
Two of the CNVs found in our cohort, both deletions, were classified as incidental findings related to NDDs. The first CNV was a 203 kb deletion within the DMD gene (hg18, chrX: 31598556-31801270) (in-frame, exons 49–53 (isoform ENST00000357033), c.7099-?_7872+?del) in a 4-year-old male patient with developmental delay, ptosis and some other facial features, but no sign of muscular dystrophy and normal muscle enzymes. Despite the recent report of X-linked ID in a family with a 3 bp DMD deletion affecting the Dp71 isoform without muscular dystrophy,45 the deletion in our patient does not affect this isoform and is less likely to explain his phenotype. Therefore, it was considered as an incidental finding with prognostic value for the patient.
The second CNV was a 380 kb maternally inherited deletion encompassing 10 genes (hg18, chrX: 70006030-70385683) in a female patient later diagnosed with a truncating mutation in ASXL1 confirming the clinical diagnosis of Opitz-Bohring syndrome.46 However, the deletion contained several known X-linked recessive disease genes and the patient, carrier mother and grandmother showed 98% skewing of X-inactivation. Therefore, it was considered as an incidental finding with predictive value of pathogenicity in males for future pregnancies.
Discussion
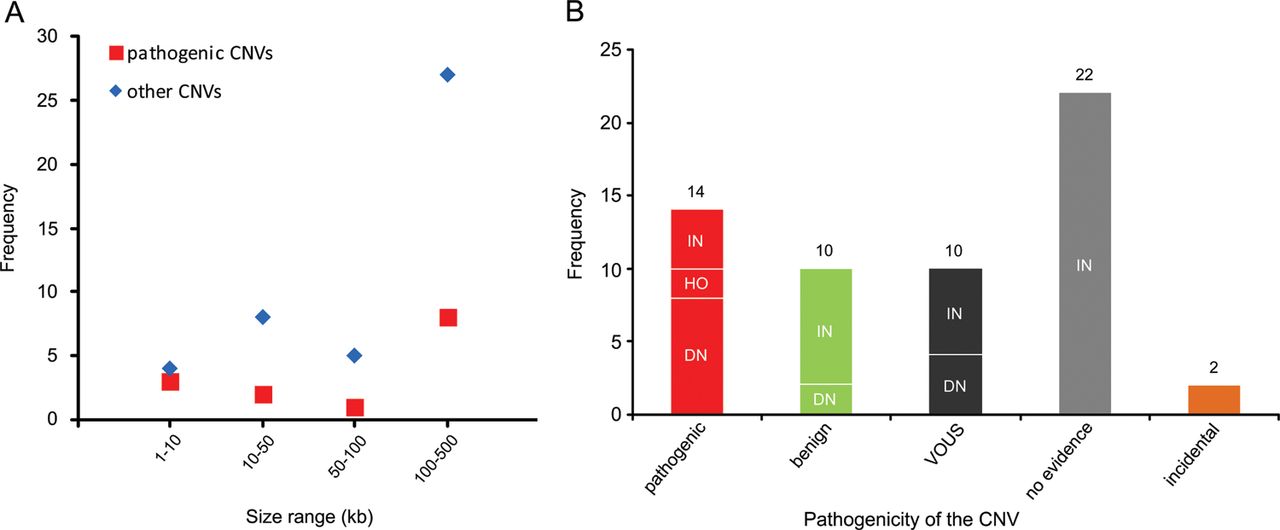
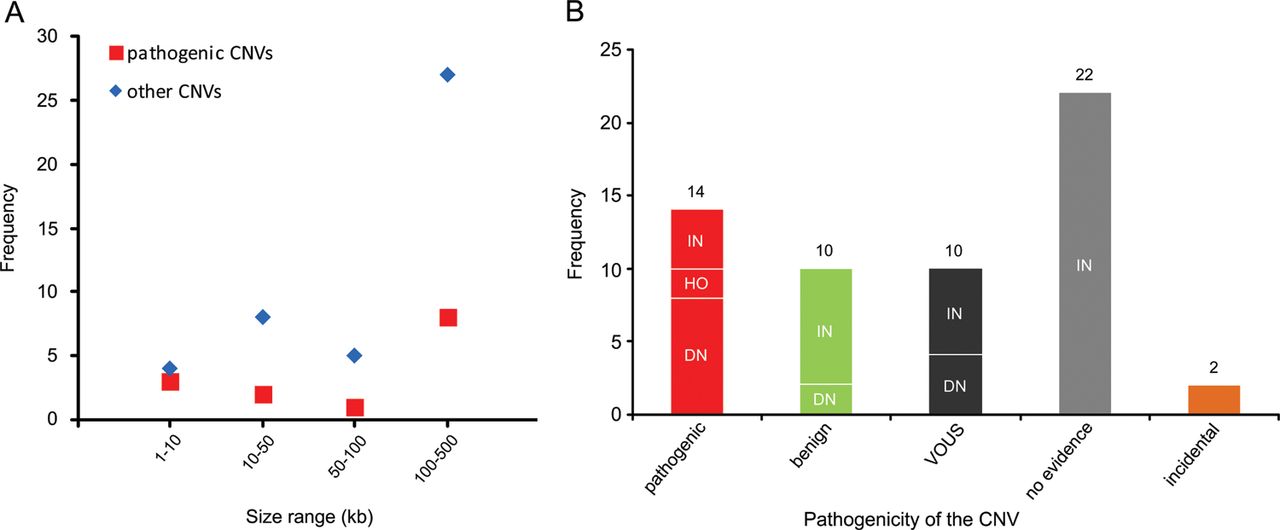
Recent genome-wide studies have shown a significant increase in the burden of rare exonic CNVs in patients with autism spectrum disorder (ASD) compared with controls7 ,8 and global burden of rare genic deletions of <500 kb compared with all CNVs,47 but did not investigate the clinical significance of individual rare CNVs. In this study, we investigated all rare exonic CNVs sizing 1–500 kb detected by genome-wide high-resolution CMA for genetic diagnosis and gene discovery in a large cohort of 714 clinically well-characterised patients with NDDs. 60.4% of such CNVs were confirmed by secondary testing and as expected, false positive aberrations were significantly smaller (median 19 vs 131 kb) in size. However, three out of seven true CNVs sizing 1–10 kb, but only 8 out of 35 sizing 100–500 kb were pathogenic, indicating the highest fraction of pathogenic CNVs in the smallest size range (figure 4A). Therefore, genome-wide exon-level CNV testing would be desirable and may be achievable by next-generation sequencing in the near future.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Distribution of copy number variants (CNVs) <500 kb in different size ranges and categories. (A) Frequency of pathogenic or likely pathogenic CNVs (pathogenic) versus other CNVs in four size ranges is shown. (B) Frequency of CNV inheritance pattern in five categories: pathogenic or likely pathogenic (pathogenic), likely benign (benign), variants of uncertain significance (VOUS), CNVs with no evidence in favour or against their pathogenicity (no evidence), and incidental findings related to NDDs (incidental). De novo or likely de novo CNVs are indicated as DN, inherited or likely inherited as IN, and homozygous as HO.
Although both intragenic deletions and duplications can lead to out-of-frame defects and gene haploinsufficiency, frequency of intragenic deletions appears to be higher than duplications. A study with targeted exon-level CNV analysis in 3018 patients with suspected Mendelian disorders illustrated a CNV detection rate of 3.3% of which 96 were deletions and only 2 were duplications.48 Recent data from WES have also indicated the enrichment of 1–30 kb deletions in individuals with ASD.7 Accordingly, 12 of our 41 confirmed rare small deletions (29%) were pathogenic, but only 2 of 17 confirmed rare duplications (12%) were categorised as such, but the difference did not reach statistical significance.
With reference to inheritance pattern (figure 4B), 24% of confirmed small CNVs were de novo or likely de novo, but only 57% of these were considered disease causing. Rare CNVs occurring de novo are more likely to be pathogenic, and consensus guidelines suggest de novo CNVs to be considered for causality of the abnormal phenotype.4 However, there are few reported instances in which candidate pathogenic de novo CNVs <500 kb eventually appeared to be benign, indicating that their causality should not be overestimated.49 Likewise we found 2 of 14 (14%) de novo rare CNVs <500 kb to be likely benign because of the identification of pathogenic mutations in known disease genes by WES in the corresponding patients, fully explaining their phenotypes (table 1; patients 62848 and 45333). For the two pathogenic de novo CNVs affecting novel disease loci, overlapping CNVs in patients with similar phenotypes were identified via the DECIPHER database. While we described in detail the novel syndrome associated with MED13L haploinsufficiency elsewhere,13 the phenotype of borderline low IQ with or without autistic features or developmental delay associated with CTNND2 deletions is first described here.
In our cohort, 71% of confirmed small CNVs were heterozygous and familial. Recent studies indicated enrichment of inherited CNVs in patients with mild clinical phenotype50 and sporadic ASD cases,8 as well as lower cognitive performance in controls carrying rare CNVs.51 However, clinical interpretation of such CNVs in individual cases remains challenging. We found four inherited heterozygous deletions (10% of confirmed familial heterozygous CNVs) to be pathogenic or likely pathogenic based on the parent's phenotype and/or reported cases in the literature. Notably, eight (20%) of the inherited small CNVs could be categorised as likely benign after finding clearly pathogenic point mutations in the patients or observing similar CNVs in new control data (see online supplementary table S2) and the rest remained without clear evidence (see online supplementary table S1; figure 4B).
In addition, our findings underline the importance of homozygous disease causing CNVs <500 kb (∼0.3% of total cohort) in a genome-wide evaluation. Pathogenic homozygous CNVs are more commonly described in consanguineous families, but limited data are available on the genome-wide estimate of these CNVs with a reported frequency of ∼0.009% in a cohort of diverse clinical phenotype52 and ∼0.5% in 194 patients with ASD.53 Here, both of our homozygous deletions were detected in patients of non-consanguineous parents of Swiss origin, the homozygous 2p21 deletion at the border of a 7.6 Mb loss of heterozygosity (LOH) region37 and the ACOT7 exonic homozygous deletion flanked by two heterozygous single-nucleotide polymorphism within a 1.7 Mb interval. CNVs or mutations affecting ACOT7 have not been reported before; however, given the familial segregation and the overlap with the reported KO mice phenotype, we suggest that ACOT7 indeed causes a novel autosomal-recessive disorder characterised by mild ID, epilepsy and episodes of ravenousness and fatigue.
In our cohort, the diagnostic yield of larger pathogenic CNVs sizing 500 kb–10 Mb was 8.8%, which is slightly lower than reported data and may be explained by the clinical preinvestigation of all patients, which allowed 13 cases with recognisable microdeletion syndromes to be diagnosed by targeted testing (deletion 22q11.2, Williams–Beuren syndrome, Smith–Magenis syndrome). These diagnoses, which are frequently reported in CMA studies, would have added another 1.8% to this cohort seen in our genetic clinic and we also excluded patients with potentially cytogenetically visible CNVs larger than 10 Mb. The overall added value of rare CNVs <500 kb to the diagnostic yield was ∼2% (1.1% de novo, 0.3% homozygous, 0.6% inherited). Of note, 79% of this diagnostic yield represented CNVs overlapping with known disease loci while 21% affected novel loci and were contributory to delineation of novel disease entities in the course of this study. In addition, two of the confirmed small CNVs (0.3% of patients) represented incidental pathological findings not related to the patient's current phenotypes. Our finding of 11 out of 714 patients (1.54%) with small pathogenic CNVs in known disease loci is higher than the 0.4–1% observed in previous studies investigating more than 300 patients each using lower resolution Agilent 44k, 105k, 180k, 244k arrays or custom-designed exon-targeted arrays.54–57
In summary, including the 0.4% of patients with pathogenic CNVs in novel disease loci, our results verify the diagnostic relevance (∼2%) of genome-wide rare CNVs <500 kb and their inherent potential to discover new conditions enabling better characterisation of NDDs.
Acknowledgments
We sincerely thank the affected individuals and their families for participation and the DECIPHER Consortium for their collaboration. This research was supported by grants from the Swiss National Science Foundation (SNF 320030_135669), Forschungskredit of the University of Zurich (grant number 54220201) and radiz–Rare Disease Initiative Zurich, clinical research priority program, University of Zurich.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All authors are justifiably credited with authorship, according to the authorship criteria.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Kantonale Ethikkommission Zurich.
-
Provenance and peer review Not commissioned; externally peer reviewed.