Article Text

Abstract
Background Mutations in Ras/mitogen-activated protein kinase (Ras/MAPK) pathway genes lead to a class of disorders known as RASopathies, including neurofibromatosis type 1 (NF1), Noonan syndrome (NS), Costello syndrome (CS), and cardio-facio-cutaneous syndrome (CFC). Previous work has suggested potential genetic and phenotypic overlap between dysregulation of Ras/MAPK signalling and autism spectrum disorders (ASD). Although the literature offers conflicting evidence for association of NF1 and autism, there has been no systematic evaluation of autism traits in the RASopathies as a class to support a role for germline Ras/MAPK activation in ASDs.
Methods We examined the association of autism traits with NF1, NS, CS and CFC, comparing affected probands with unaffected sibling controls and subjects with idiopathic ASDs using the qualitative Social Communication Questionnaire (SCQ) and the quantitative Social Responsiveness Scale (SRS).
Results Each of the four major RASopathies showed evidence for increased qualitative and quantitative autism traits compared with sibling controls. Further, each RASopathy exhibited a distinct distribution of quantitative social impairment. Levels of social responsiveness show some evidence of correlation between sibling pairs, and autism-like impairment showed a male bias similar to idiopathic ASDs.
Conclusions Higher prevalence and severity of autism traits in RASopathies compared to unaffected siblings suggests that dysregulation of Ras/MAPK signalling during development may be implicated in ASD risk. Evidence for sex bias and potential sibling correlation suggests that autism traits in the RASopathies share characteristics with autism traits in the general population and clinical ASD population and can shed light on idiopathic ASDs.
- Autism
- Neurofibromatosis Type 1
- Costello Syndrome
- Noonan Syndrome
- Cranio-Facio-Cutaneous Syndrome
Statistics from Altmetric.com
Introduction
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterised by specific impairments in language, communication, social skills and presence of restricted or repetitive interests and behaviours. The prevalence of ASDs is estimated to be 0.5–2.0% in the USA.1 ,2 It has been long known that several Mendelian disorders are associated with autism. The most well established of these, including fragile X syndrome, tuberous sclerosis, Rett syndrome, and PTEN mutation account for up to 5% of ASDs.3 Additional rare autosomal dominant or recessive disorders, such as Smith–Lemli–Opitz syndrome, Timothy syndrome and CHARGE syndrome have been described as associated with autism in clinical reports.4 There is recent resurgence of interest in rare highly penetrant single nucleotide variants (SNV) analogous to these traditional genetic models that may influence risk for idiopathic autism.5–9 To date, estimates suggest that SNVs and copy number variants (CNV) acting in dominant, recessive, or X-linked models might account for a small proportion of autism, and common single nucleotide polymorphisms (SNP) can account for nearly half the variation in autism, with the remainder yet unknown, but potentially including additional, less penetrant rare variants, or complex mechanisms, such as gene–gene interaction or gene–environment interaction.10 However, it is yet unclear whether the same genes may act through rare, highly penetrant mutations and common genetic risk factors. Additionally, although many approaches have attempted to identify ‘pathways’ implicated in ASDs to unify disparate genes, these data have not converged to provide conclusive and well-replicated evidence.
There is emerging speculation that dysregulation of Ras/mitogen-activated protein kinase (Ras/MAPK) signalling contributes to common risk factors and rare known genetic causes of ASDs.11 The Ras/MAPK pathway is best known for somatic mutations in cancer, but its signalling is essential in the regulation of the cell cycle, differentiation, growth and cell senescence, all of which are critical in development. Genome-wide CNV analyses suggest Ras/MAPK pathway involvement in idiopathic autism.12 SNP association and rare sequence variants in the Ras/MAPK pathway in ASDs have been reported.13 ,14 A recent review summarises ASD candidate genes and/or CNVs related to Ras/MAPK signalling.15 Intersection with the Ras/MAPK pathway is also found in syndromic disorders with high penetrance of ASDs, including tuberous sclerosis, Smith–Lemli–Opitz syndrome, Rett syndrome, and fragile X syndrome.16 However, these prior reports are inferential at best in ascribing a relationship between Ras/MAPK activity and ASDs, and no previous study has systematically assessed individuals with germline mutations along multiple direct members of the classical Ras/MAPK signalling pathway for autism traits.
Germline mutation in genes that encode primary protein components of the Ras/MAPK pathway causes a class of developmental disorders called RASopathies (table 1). Initial reports suggested an increased rate of autism in individuals with the most common of the RASopathies, neurofibromatosis type 1 (NF1),17–19 although conflicting data called these reports into question.20 ,21 Recent studies have supported an association using questionnaire measures of dimensionally assessed autism-relevant traits.22 ,23 In addition to NF1, other RASopathies include Noonan syndrome (NS), Costello syndrome (CS), and cardio-facio-cutaneous syndrome (CFC), and together they make up a common class of single-gene developmental disorders. While each RASopathy exhibits unique phenotypic features, they share characteristic features including craniofacial dysmorphology, cardiac malformations and cutaneous, musculoskeletal and ocular abnormalities, as well as varying degrees of neurocognitive impairment, usually none to mild in NF1 and NS, and anywhere from mild to severe in CS and CFC.24–28 The literature or parent report, to date, suggests features reminiscent of autism to some extent for each RASopathy, but is far from conclusive.29–35
Four RASopathies of the study population
Although similarities in the cognitive and behavioural profiles of RASopathies and ASDs can be extracted, no previous systematic assessment of ASDs or autism traits across the RASopathies has been performed. In NF1, overall cognitive deficits have been associated with specific brain lesions (T2 hyperintensity on MRI), thus attributed to secondary effects rather than primary consequences of increased Ras/MAPK activity.36 Studying additional RASopathies not characterised by these lesions can aid in identifying true ‘pathway’ effects.37 ,38 Additionally, no data exists separating RASopathy mutation effects from shared genetic and environmental similarities with unaffected siblings, or comparing RASopathy autism trait profiles with a clinical ASD sample. We therefore screened a large RASopathy population for autism traits and compared them with unaffected sibling controls and idiopathic ASD subjects to determine the prevalence of autism features in each of the RASopathies individually and whether, as a class, these disorders might shed light on genetic risk for ASDs. This study is the first of its kind and can thus provide unique and important insight into the role of a well-known signalling pathway in predisposition to ASDs.
Subjects and methods
UCSF subjects
Subjects with RASopathies were recruited at the University of California, San Francisco (UCSF) NF/Ras Pathway Genetics Clinic and three national RASopathy family meetings (Chicago, Illinois, USA, July 2011; Berkeley, California, USA, July 2009; Orlando, Florida, USA, August 2013). Additional families were recruited at a UCSF NF Symposium (November 2011), and through RASopathy groups: NF, Inc., Children's Tumor Foundation, Noonan Foundation, CFC International, Costello Syndrome Family Support Network, and Costello Kids. Inclusion criteria was reported diagnosis of CFC, CS, NS, or NF1 by a medical geneticist, or diagnosis of NF1 by a neurologist. RASopathy subjects recruited at the UCSF clinic had initial or confirmed diagnoses by a medical geneticist (KAR). NF1 diagnosis was made using well-established criteria specified by the National Institutes of Health (NIH) Consensus Development Conference.39 Diagnosis of NF1, CFC, CS and NS subjects recruited via meetings or RASopathy groups was by parent report, and diagnosis by molecular testing was reported for a subset of all NF1 (11%), NS (56%), CS (84%) and CFC (83%) subjects. All subjects had parents with fluency in English. Because not all subjects were diagnosed by study personnel or reported a known mutation, we performed main analyses with and without individuals with reported mutations, and determined that inclusion of non-molecular diagnoses did not have a significant impact on our results (data not shown). Questionnaires were distributed in person or by mail.
Of 231 RASopathy probands, 108 have healthy full sibling participants. Siblings of RASopathy subjects were excluded if there was parental indication of possible RASopathy or ASD reported prior to data collection. This included two siblings with suspected NF1, and three siblings with ASDs. Two CFC probands in two different pairs of monozygotic twins were excluded. Participants less than 12 months old (n=1) were excluded, since questionnaire items pertain to behaviours typically exhibited above this age. Participants less than 3 years old were excluded if more than two questionnaire items were blank (n=2), as this was a potential indicator that pertinent developmental milestones had not been mastered. Subjects with idiopathic ASDs were recruited at the UCSF Autism Clinic and through local outreach, including the Golden Gate Regional Center. All ASD subjects had received prior ASD diagnosis by a professional and had no known autism-associated genetic disorders. All participants provided written consent as approved by the UCSF Human Research Protection Program (CHR #10-02794).
UCLA subjects
At UCLA, participants with NF1 aged 10–45 years were recruited via online posting (NF, Inc., and Children's Tumor Foundation), as well as through the Neurofibromatosis and Neurocutaneous Disorders clinic at Children's Hospital, Los Angeles. All had been previously diagnosed with NF1 by a physician. Prior to entering the study, a study paediatric neurologist (TR) confirmed that all NF1 participants fulfilled the diagnostic criteria specified by the National Institutes of Health Consensus Development Conference. Additional inclusion criteria required that the subject be fluent in English, have a full-scale IQ greater than 70 with no history of MR, and no significant intracranial pathology (brain tumour, hydrocephalus) indicated by reports of previously diagnosed MRI abnormalities. Because these inclusion criteria were slightly different from the UCSF criteria (IQ, MRI screening), main analyses were compared in the UCLA and UCSF components of the NF1 sample separately, and no significant differences were noted (data not shown). All participants provided written consent for participation, as approved by the institutional review board of the University of California, Los Angeles (UCLA, IRB#10-000518).
Questionnaire measures
Social Communication Questionnaire
The Social Communication Questionnaire (SCQ),40 based on the Autism Diagnostic Interview,41 is used as a binary measure of autism traits, with the threshold of scores 15 or higher used as a proxy for significant autism features.42 When possible, the SCQ was given to parents of RASopathy subjects to fill out pertaining to their affected child and one healthy sibling. This screening instrument has established validity for an autism diagnosis, with a sensitivity of 86% and specificity of 78% when distinguishing autism from other diagnoses (excluding intellectual disability), and a sensitivity of 97% and specificity of 73% when distinguishing autism from intellectual disability.42 Previous reports indicate high base rates of Attention Deficit Hyperactivity Disorder (ADHD) in NF1,22 ,23 but studies indicate that the measure of ASD traits by SCQ is unaffected by ADHD symptomatology in clinical and general population samples.43 ,44
Social Responsiveness Scale
The Social Responsiveness Scale (SRS) was distributed with the SCQ to parents of RASopathy subjects.45 ,46 The SRS is a 65-item parent questionnaire that measures social deficits characteristic of autism on a continuous severity index based on gender-normalised transformed scores (T-scores). In a normative sample, individuals with mild to moderate autism typically score in a range of 60–75, and severe autism is described at 76 or higher. When distinguishing severe autism from non-autism, the sensitivity and specificity of the SRS are 78% and 67%, respectively.41 In addition to a total SRS score, subscores are given in five domains: social communication, social motivation, social awareness, social cognition and autistic mannerisms (restricted interests and repetitive behaviours). Previous studies have indicated that the severity of autism traits measured by the SRS is highly heritable in clinical populations and in the general population. Data obtained from families including at least one autistic child report correlation for parent-report SRS scores 0.35–0.38.47 ,48 Similar SRS correlations (0.37–0.63) were found in samples of non-autistic fraternal twin pairs.49 ,50 Further, SRS scores are not significantly correlated with IQ, and not affected by the presence of ADHD.45 ,49 ,51–53
Because we included some subjects younger than the ages for which these instruments have been validated, analysis was performed with and without the subjects younger than 3 years old (SCQ) or 4 years old (SRS) and the results were not different (data not shown).
Adult participants without available parents to complete the SCQ and SRS (n=22) were given the SRS for Adults (SRS-A) to be filled out by someone well acquainted with the participant, such as a spouse or sibling. Each item in the SRS-A corresponds to an item in the child and adolescent SRS, with slight variations in content that are developmentally appropriate for adults.51 Transformed scores from the SRS-A can thus be directly compared with parent-report SRS, and have been combined here.
In-person assessment
Local subjects to the San Francisco Bay Area who participated in our study and scored at or above the ASD cutoff for SCQ, SRS, or both (and one negative control scoring in the typical range on both), were offered participation in more extensive in-person assessment (n=34). Those who agreed to return for validation were administered comprehensive autism diagnostic testing (n=7) using the Autism Diagnostic Observation Schedule-Second Edition Generic (ADOS), Autism Diagnostic Interview, Revised (ADI-R), Vineland Adaptive Behaviour Scales, Second Edition Parent/Caregiver Rating Form (VABS-II), see below. IQ testing was performed using age-appropriate Wechsler IQ tests.54 ,55 Additionally, a child psychiatrist met with subjects. A consensus panel discussed each validation subject and reviewed the research instruments and clinical impression. This panel included a child psychiatrist (RH), a psychologist (SR), a human geneticist (LAW), a child neurologist (EJM), a medical geneticist specialising in RASopathies (KAR), and the certified individual who administered the testing (FW).
Autism diagnostic observation schedule
The ADOS is a 30 min semistructured interview that assesses social interaction, communication, play and imaginative use of materials for individuals who may have autism.56 We used two modules of the ADOS Second Edition adapted to verbal fluency in children (module 3) and adolescents or adults (module 4). The ADOS has been shown to give reliable autism diagnoses in mixed samples with ASDs, other psychopathologies, intellectual disability and typical development,57–60 with a sensitivity of 92% and a specificity of 65% when distinguishing autism from non-autism and a sensitivity of 87% and specificity of 47% when distinguishing an ASD from non-autism.58 ADOS ranks items in domains of social behaviour, communication and restricted or stereotyped interests on a scale from 0 (not abnormal) to 2 (abnormal) or 3 (severely abnormal). There is a classification of either autism or ASD when scores meet or exceed the cutoffs for social behaviour, communication and behaviour and communication combined. The ADOS was conducted and scored by research-certified examiners, and sessions were videotaped to conduct further analysis during the consensus process.
Autism diagnostic interview-revised
The ADI-R61 is a 93-item standardised interview given to a parent or caregiver to assess autism traits according to the DSM-IV criteria. Scoring algorithms based on age yield transformed scores in reciprocal social interaction, communication and restricted, repetitive and stereotyped behaviours. It is often used in conjunction with the ADOS because it assesses developmental and behavioural abnormalities that are beyond the scope of the ADOS, which captures spontaneous social behaviours in a limited number of planned activities. Unlike the ADOS, it provides score cutoffs for autism but not ASDs. An autism classification is given when subjects receive scores above the cutoff in all three ADI-R domains. Sensitivity and specificity in a sample of toddlers were 92% and 72%, respectively.62 In a sample aged 5–20 years old, sensitivity and specificity were 77% and 63%.58
Vineland Adaptive Behaviour Scales II
The VABS take the form of a semistructured parent interview, which we used to evaluate three areas of adaptive behaviour: communication, daily living skills, and socialisation.63 VABS has been used to distinguish children with autism from age-matched and IQ-matched children with non-autistic developmental disorders.64 Although VABS is affected by IQ, Kraijer et al indicated that individuals with autism exhibit a pattern of adaptive deficiencies: extreme deficits in socialisation, relative strengths in daily living skills, and intermediate scores in communication.65 ,66 We used the VABS (Second Edition) to broadly assess the adaptive abilities of the RASopathy subjects relative to their age group.
Data analysis
To assess the prevalence of autism-like impairment by group (eg, RASopathy, control, ASD, genders), a one-sided Fisher's Exact test was used to compare the proportion of above-threshold SCQ scores, as our expectations were unidirectional (RASopathy subjects show increased proportion, males show increased proportion). The SRS T-score distributions were compared using two-sided F-tests for variance and Student t tests for means. One-sided t tests were used to compare mean SRS scores between male and female cases, and to compare RASopathy, ASD and sibling subjects. Two-sided t tests were used to compare mean SRS scores between different RASopathies. All main analyses were conducted with and without related individuals (eg, comparison of NF1 probands (one per family) to only CS/CFC/NS siblings to exclude any related pairs), in order to confirm that inclusion of genetic relatives did not bias our results (data not shown). Because our analyses compared each RASopathy to controls, to account for multiple testing across the four RASopathies, we consider p<0.01 to be criteria for significance and 0.05>p>0.01 to be suggestive.
To assess whether the SRS subscore profiles were different between RASopathy and idiopathic ASD subjects who were in the ‘affected’ category of the SCQ, we performed a Principal Components Analysis (PCA) to see whether five symptom domain subscales of the SRS (social awareness, social cognition, social communication, social motivation and autistic mannerisms) distinguished RASopathy from ASD subjects. The PCA was run in R (R: A language and environment for statistical computing, Vienna, Austria) using the dudi.pca function from the ade4 package.67 ,68
To assess whether shared genetics influenced the severity of autism traits within families, we tested for a correlation between SRS scores of RASopathy subjects and their matched healthy siblings (one pair per family). SRS scores were normalised to the mean in each group (NF1, CS, NS, CFC and unaffected siblings), and Pearson coefficients and one-tailed p values were calculated for sibling pairs, as we would only expect positive sibling correlation.
Results
Study population characteristics
A total of 521 individuals completed either the SRS (n=60), the SCQ (n=22), or both (n=439). These included 81 NF1, 44 CS, 52 NS, 54 CFC, 171 ASD subjects and 119 unaffected siblings of RASopathy subjects. There were no significant differences in the number of male and female participants except in ASD (73% male, p<0.001, see online supplementary table S1). Ages were not significantly different between RASopathy subjects, siblings and ASD subjects. In the CS sample, the most common HRAS mutations were Gly12Ser (59.1%) and Gly13Cys (15.9%). No genetic testing results were available for 15.9% of CS subjects. Of CFC subjects, 54.7% had BRAF mutations, 9.4% had mutations in MAP2K1, 11.3% had mutations in MAP2K2 and 1 individual (1.9%) had a KRAS mutation. No genetic testing results were available for 24.5% of CFC subjects. NS subjects had mutations in PTPN11 (34.6%), SOS1 (9.6%), SHOC2 (3.9%), KRAS (3.9%) and RAF1 (3.9%). No genetic testing results were available for 41.2% of NS subjects. The majority of NF1 subjects were clinically diagnosed and, therefore, did not report genetic mutations. Only 12.3% of NF1 subjects reported known mutations, and no single mutation was reported in more than two unrelated individuals.
Prevalence of autism traits by the SCQ
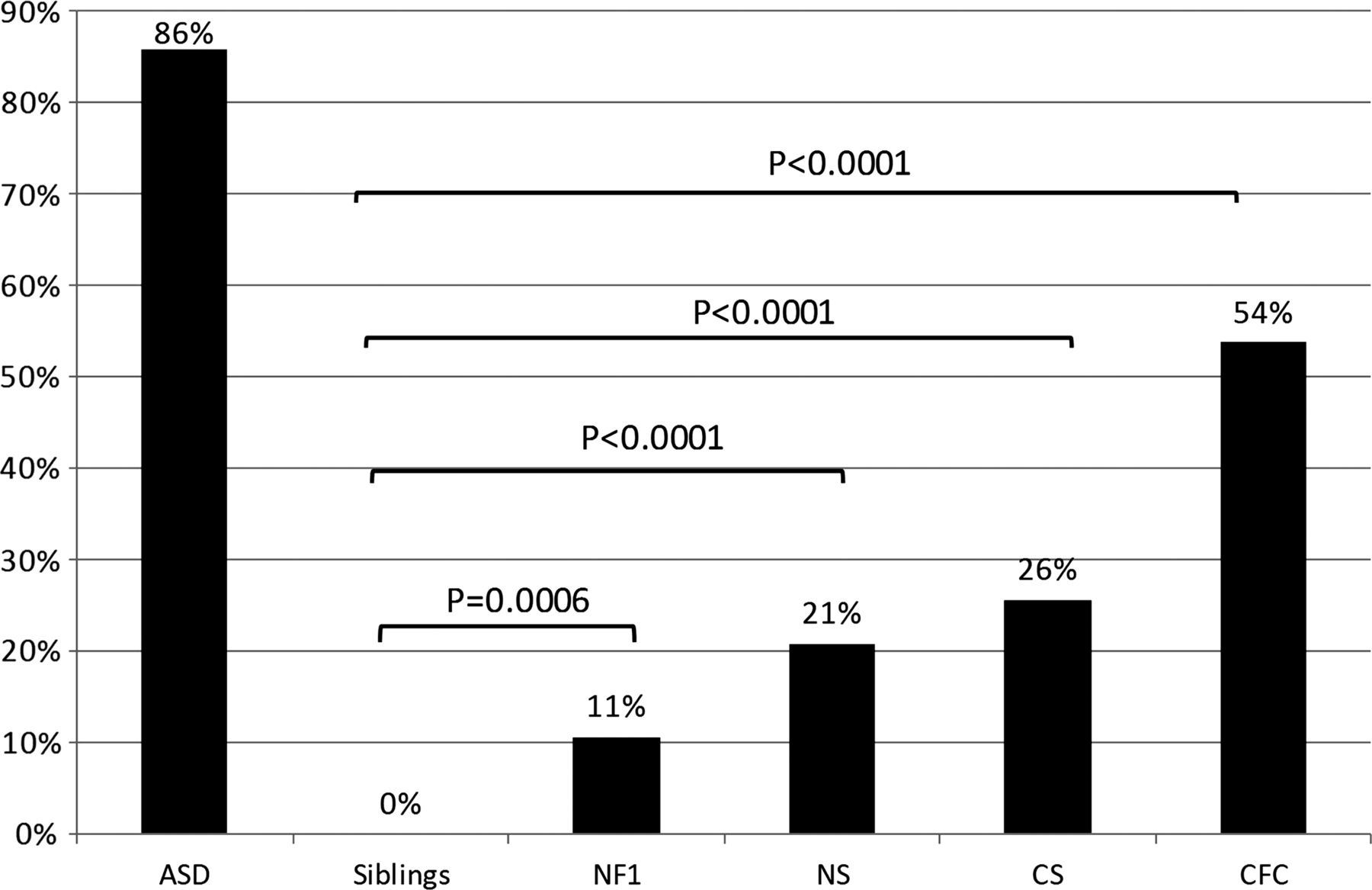
SCQs were conducted for 461 individuals with ASDs (n=133), NF1 (n=66), NS (n=48), CS (n=43), CFC (n=54), and unaffected siblings of RASopathy subjects (n=117), and results are summarised in figure 1. In our sample of clinically ascertained ASD subjects, 85% scored at the threshold of 15 or higher, consistent with the reported SCQ sensitivity of 86–88%.41 ,42 The SCQ has a reported specificity of 72–78%.41 ,42 No siblings unaffected with a RASopathy met the threshold of 15 on the SCQ. When RASopathy subjects (n=213) were considered together, 27% of the total sample scored above the threshold (compared to siblings, p<0.0001). However, above-threshold SCQ scores were not evenly distributed between RASopathies: 54% of CFC subjects, 26% of CS subjects, 21% of NS subjects, and 11% of NF1 subjects had SCQ scores above the threshold indicative of autism. The proportion of individuals meeting SCQ screening criteria for autism was significantly higher than sibling controls for CFC, NS and CS (p<0.0001), and for NF1 (p=0.0006).

Social Communication Questionnaire (SCQ) in the RASopathies. Percentage of subjects with SCQ scores of ≥15 for individuals with an idiopathic autism spectrum disorder (n=133), unaffected siblings of RASopathy probands (n=117), neurofibromatosis type 1 (NF1) (n=66), Noonan syndrome (NS) (n=48), Costello syndrome (CS) (n=43), and cardio-facio-cutaneous syndrome (CFC) (n=54). One-tailed Fisher's Exact test p values were calculated as indicated.
Distribution of autism traits by the SRS
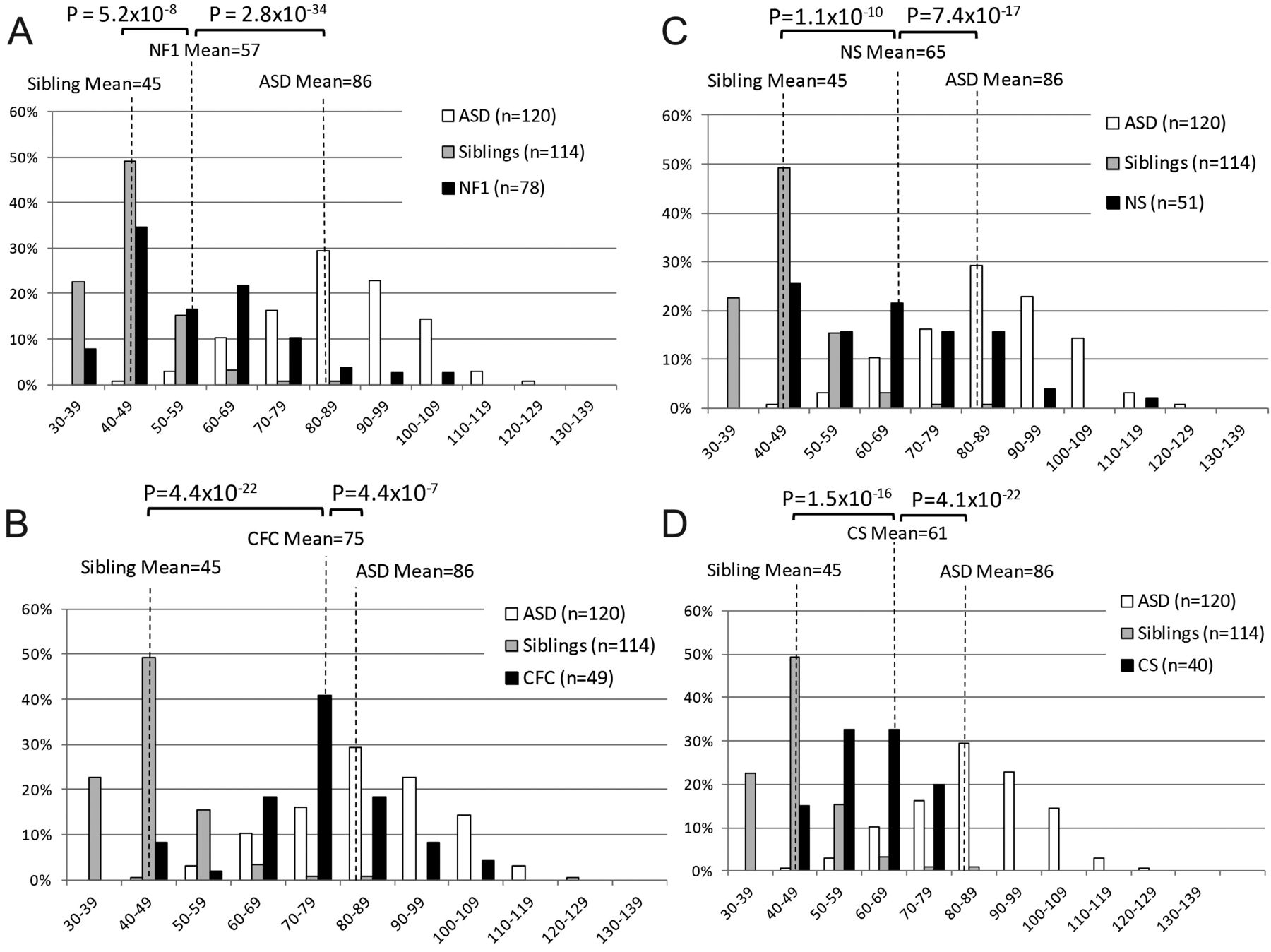
The SRS was used to examine the distribution of autism traits within and across RASopathies, as it provides a quantitative normalised score. There were SRS data for 167 ASD subjects, 218 RASopathy subjects (78 NF1, 51 NS, 40 CS and 49 CFC) and 114 unaffected siblings. ASD subjects had an average transformed SRS score (T-score) of 86 (SD=14); this is consistent with SRS screening criteria for clinically significant social impairments (indicated by T-scores ≥60) and for autism (T-score ≥76). RASopathy subjects combined had an average T-score of 63 (SD=16), which was significantly lower than idiopathic ASD T-scores (p=1.8×10−38) and significantly higher than the average T-score of unaffected siblings (46, SD=10, p=4.5×10−32). The greatest impairments were seen in CFC subjects (mean=74, SD=13, figure 2D), followed by NS (mean=65, SD=17, figure 2C), CS (mean=61, SD=10, figure 2B), and NF1 (mean=57, SD=16, figure 2A). The average T-score for ASD subjects was significantly different than CFC (p=4.4×10−7), NS (p=7.4×10−17), CS (4.1×10−22), and NF1 (2.8×10−34) (figure 2). The average sibling T-score was significantly lower than all RASopathies: CFC (p=4.4×10−22), NS (p=1.1×10−10), CS (p=1.5×10−16), and NF1 (p=5.2×10−8). Comparison of the mean T-scores within RASopathies showed that scores for CFC subjects were significantly different (p≤0.0018) than each of the other three RASopathies. NF1 and NS had borderline different SRS T-scores (p=0.011).
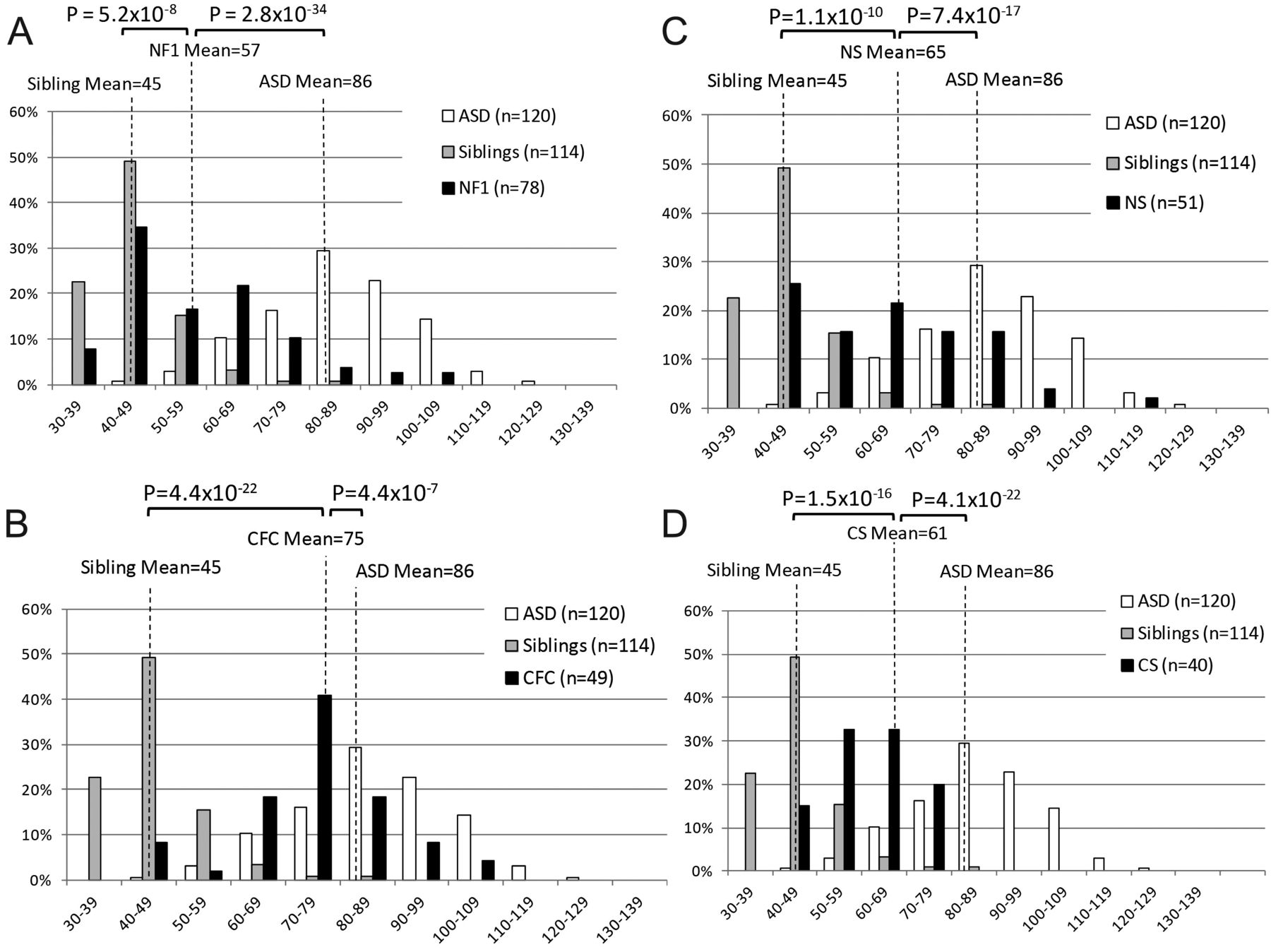
Social Responsiveness Scale (SRS) in the RASopathies. Histograms depicting the distributions of transformed SRS scores for idiopathic autism spectrum disorders (ASDs), unaffected siblings of RASopathy probands, and (A) neurofibromatosis type 1 (NF1), (B) Costello syndrome (CS), (C) Noonan syndrome (NS), and (D) cardio-facio-cutaneous syndrome (CFC). Distributions are given as the percentage of individuals who scored within each bin range. One-tailed p values from a Student t test comparing average T-scores of RASopathy subjects, idiopathic ASD subjects, and unaffected siblings of RASopathy probands are indicated.
The distribution of SRS scores within each RASopathy is distinct if variance is taken into account. While means can identify average differences across populations, comparison of variance can have implications about the distribution of scores around the mean. For example, two RASopathies could show a similar mean in the mild to moderate range of the SRS, with one group showing a tight unimodal distribution around this mean and the second showing a bimodal distribution with a subset similar to controls and a subset similar to ASDs. These might imply different genetic mechanisms, such as a main effect of a RASopathy mutation (unimodal) or a epistasis (bimodal). Variances were not significantly different between CFC and NS (p=0.11), CS (p=0.07), or NF1 (p=0.11), or between NF1 and NS (p=0.68), the pairs for which the means were different. However, NF1 and NS both had significantly higher variance than CS (p<0.001) and unaffected siblings (p<4.3×10−10). The SD in CFC scores (13) was not significantly different from ASDs (14, p=0.61), but was significantly higher than unaffected siblings (9, p=0.0002, figure 2D). The variance of CS scores was suggestively lower than ASD subjects (p=0.015) but not significantly different than unaffected siblings (p=0.19, figure 2B). However, it should be noted that in unaffected siblings, there is a ‘basement’ effect where the distribution is cut off by a lower bound.
SRS scores by SCQ threshold
To determine whether the SRS score distribution within the RASopathies was bimodal (eg, consisting of subpopulations similar to unaffected siblings and similar to ASD subjects), we analysed the average SRS T-score for RASopathy subjects that were above and below the autism threshold by SCQ. When considering only ASD and RASopathy subjects with SCQ scores of 15 or higher, the mean SRS T-score for RASopathy subjects (n=53) was 79 (SD=14) and the distinction between the SRS scores of RASopathy and ASD subjects (mean=88; SD=13) was significantly lower (p=1.7×10−4). On the other hand, when considering SRS scores for RASopathy subjects (n=145) and unaffected siblings (n=112) with an SCQ score less than 15, the difference between the average T-scores of RASopathy subjects (mean=59, SD=13) and unaffected siblings (mean=45; SD=9) remained significantly higher (p=1.3×10−19).
Patterns of deficits by SRS subscales
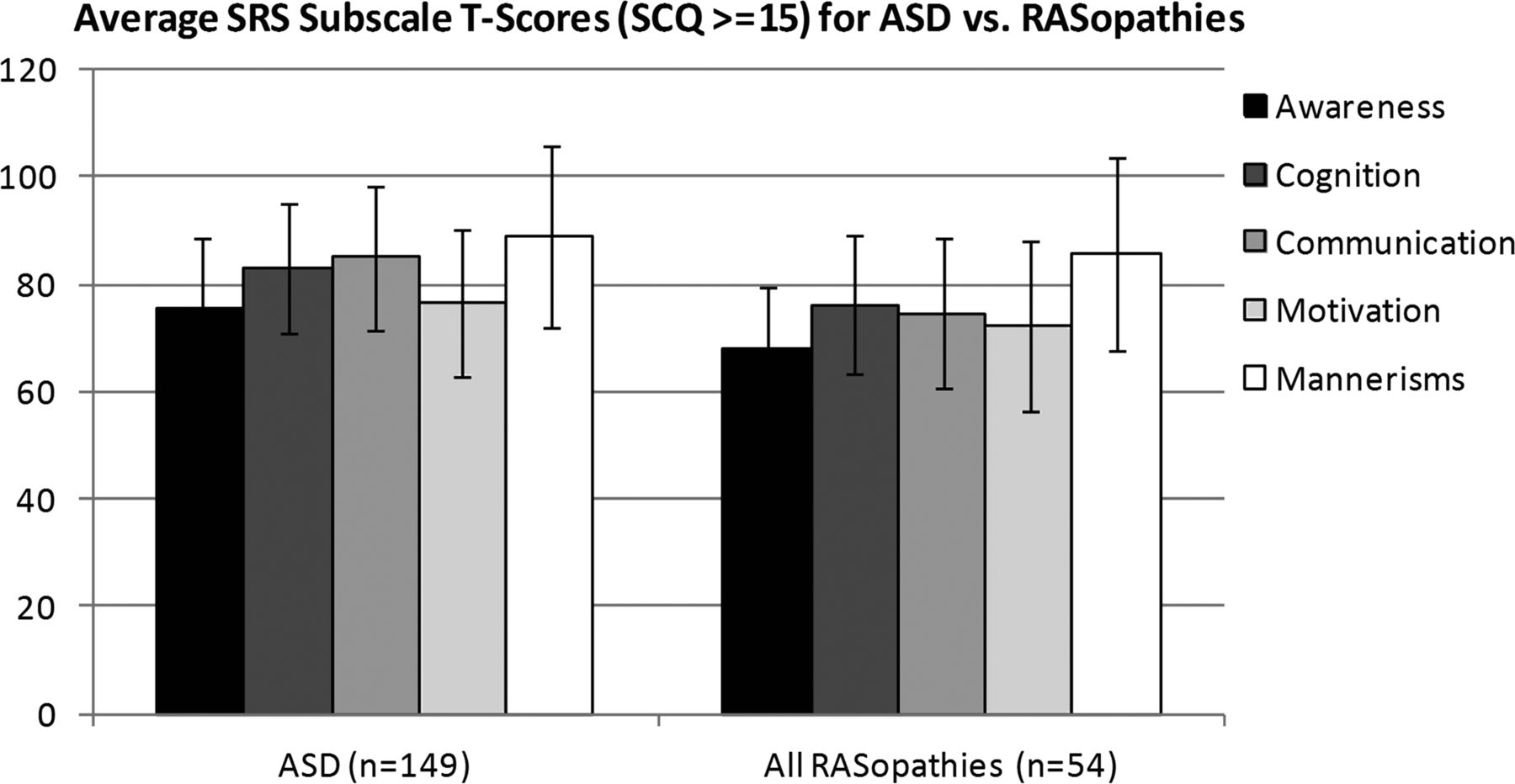
To assess whether the RASopathies and ASDs showed distinct patterns of social deficits across domains measured by the SRS, we analysed the domain subscores in social awareness, cognition, communication, motivation and autistic mannerisms for subjects who were above-threshold by SCQ. Using this analysis, we could assess whether RASopathy subjects showing significant autism traits had high scores driven by the same symptoms as subjects with idiopathic ASDs. For example, we would expect that if cognition or attention issues were driving RASopathy scores as opposed to the more classic autistic traits, the RASopathy subjects would show a distinct profile from idiopathic autism.
There were 54 RASopathy subjects with SRS scores (7 NF1, 10 CS, 10 NS, 27 CFC) and scores of 15 or higher on the SCQ. The mean subscale scores and SDs for RASopathy and ASD subjects show that both groups have impairment across the five domains in a similar pattern, thus, RASopathy SRS scores are not simply driven by impairment in a single domain (figure 3). PCA using the five SRS subscale scores demonstrates that RASopathy subjects and idiopathic ASD subjects do not cluster separately, suggesting that they were not driven by different components (figure 4). Further, this implies that relative genetic homogeneity in RASopathy subjects does not dictate significant phenotypic homogeneity compared with idiopathic ASDs by this measure.

Social Responsiveness Scale (SRS) Subscale Scores in autism spectrum disorders (ASDs) vs RASopathies. Average transformed scores in SRS subscales for all RASopathy (7 neurofibromatosis, 10 Costello syndrome, 10 Noonan syndrome, 27 cardio-facio-cutaneous syndrome) and idiopathic ASD subjects with a Social Communication Questionnaire score of 15 or higher. Error bars depict the SD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Principal Components Analysis (PCA) on Social Responsiveness Scale (SRS) Subscales in autism spectrum disorders (ASDs) versus RASopathies. The first two components of a Principal Components Analysis of the five SRS subscale scores for RASopathy and ASD subjects.
Sibling correlations
We conducted correlation analyses on the SRS scores of RASopathy subjects and their matched healthy siblings to test for similarity between siblings potentially indicating a shared genetic basis for social responsiveness measured by the SRS. It has been recently suggested that variable penetrance for quantitative traits like IQ and social responsiveness in the presence of a genetic risk factor, such as a CNV, might be primarily influenced by background genetics.69 If that were the case here, we would expect to observe sibling correlation equivalent to those previously reported for the SRS, 0.35–0.38.47 ,48 ,50 Correlations were calculated for 101 independent sibling pairs (21 NF1, 25 CS, 27 NS and 28 CFC). Transformed SRS scores were normalised to the mean SRS score for the RASopathy or for unaffected siblings, and Pearson coefficients and one-tailed p values were calculated. The highest SRS correlations were seen between the NF1 affected-unaffected sibling pairs (r=0.41, p=0.033, see online supplementary figure S1A) and CFC affected-unaffected sibling pairs (r=0.29, p=0.067, see online supplementary figure S1D), similar to previous sibling correlation, although these show only suggestive significance considering the number of comparisons we have made. However, we note that the correlations in NF1 and CFC are driven by relative outliers, making these data difficult to interpret without replication or larger sample sizes. The SRS correlation between CS probands and their unaffected siblings was positive (r=0.20, p=0.17), but lower than that of NF1 or CFC sibling pairs. The NS sibling correlation is lower than the other RASopathies (r=0.071, p=0.36), but a strong positive correlation exists when pairs with a proband scoring below (fewer autism traits) the SRS mean for NS (n=14) are considered separately (r=0.54, p=0.023, see online supplementary figure S1C).
Sex bias of autism traits in the RASopathies
The male bias in autism is evident in the population prevalence of ASDs in males and females which occurs at approximately a 4.1:1 ratio.70 The male bias in autism traits is reflected in our RASopathy sample in both SCQ and SRS scores (table 2, see online supplementary table S2). By SCQ, there was a significantly higher prevalence of above-threshold SCQ scores in males versus females women: 38% of male RASopathy subjects (n=102) and only 17% of the females (n=109) scored above threshold (p=0.0003).
Above-threshold SCQ scores in male and female RASopathy subjects and healthy siblings
Male bias of autism traits was most evident in NF1: 27% of males (n=26) and zero females (n=40) scored above the SCQ threshold (p=0.0008). The average SRS T-score in NF1 males was 61.3 (SD=20), while NF1 females had a borderline lower average score (mean=54.3, SD=14, p=0.048). Higher levels of autism traits were also seen in males for CS and CFC, although the prevalence of social impairments in males was not significantly higher for the SRS or SCQ. Male bias was least evident in NS subjects: 23% of all NS males (n=26) and 18% of NS females (n=22) scored above the SCQ threshold. The mean T-score of NS maleas was 64.9 (SD=15) compared with a mean T-score of 64.3 (SD=20) in females (p=0.46). By contrast with RASopathy probands, no gender differences in SRS and SCQ scores were seen in healthy siblings.
In-person assessment using research and clinical instruments
Subject A was an NF1 female negative control with subthreshold SRS and SCQ scores, and the in-person visit confirmed no impairment (see table 3). Subject B and Subject G were an NF1 female and an NS male with borderline SRS scores of 73 and 64 and subthreshold SCQ scores of 14 and 6, respectively. Although they each showed some distinct areas of social impairment by testing and clinical impression, neither met ADOS or ADI-R criteria for an ASD. Subjects who scored above the severe SRS cutoff of 75 (Subjects C, D, E, F) received an ADOS classification of ASD (Subject D) or autism (Subjects C, E, F). In accordance with the ADOS, the ADI-R indicated autism for Subjects C and F, and was only one point below the autism cutoff for Subject E. Subject D did not meet the ADI-R cutoffs for autism, which is consistent with the ADOS classification of ASD for this subject because the ADI-R does not have score cutoffs for ASDs other than strict autism. In these subjects, clinical impression confirmed the ADOS classification, except for Subject E, who was the only subject in this group with an SRS above 75 and a below-threshold SCQ score. Clinical impression of Subject E was that the criteria of autism were met, but the presentation was atypical compared with idiopathic autism.
Participant visit outcomes
By ADOS, the most frequent deficits in communication were speech abnormalities (intonation, volume, rhythm, rate) (n=5), limited inquiry into the thoughts, feelings, or experiences of others (n=5), and limited sharing of information about oneself (n=6). The most frequent deficits in reciprocal social interaction were poor recognition of other people's emotions (n=5) and insight into typical social relationships (n=5). Most subjects (n=5) had deficits in the imaginative use of objects or verbal descriptions.
Discussion
In the first study assessing a large cohort with germline mutations across the Ras/MAPK pathway, we demonstrate that RASopathy subjects have significant social impairment compared with sibling controls using two parent-questionnaire measures. Similar studies have been recently performed for subjects with NF1 using the SRS.22 ,23 We have expanded upon their approach by including a binary classifier questionnaire (SCQ), clinical observation of several subjects, and comparison with related RASopathies as well as sibling controls and idiopathic ASDs. However, our results by SRS for NF1 were consistent with these recent reports: the mean SRS score for the NF1 sample in both published studies was approximately 60, and about 30% received clinically relevant scores between 60 and 75. In our NF1 sample (n=78), the mean T-score was 57, and 30% received a score between 60 and 75. In our study, 12.8% received scores of 75 and above. In the Walsh et al22 study, 13% of the NF1 subjects (n=62) had scores of 75 and above, while Garg et al23 (n=109) had a higher proportion (29%) of NF1 subjects who scored within this severe range.
We observed that the fraction of clinically significant impairment varies across the RASopathies, with CFC showing the highest proportion of affected individuals and NF1 showing the lowest. Interestingly, autism traits do not track simply with the level of cognitive impairment by disorder. Only 6–7% of NF1 patients and 17% of NS patients have mild cognitive impairment,26 ,71 while there is 30% mild impairment and 50% moderate-severe impairment in CS, and 24% mild and 70% moderate-severe impairment in CFC.27 ,28 In our dataset, NS showed autism traits as severe as CS. Interestingly, NS and CFC had the highest levels of autistic traits by SRS, and the genes associated with these disorders span the entire Ras/MAPK pathway.
Social responsiveness varies within each of these highly penetrant disorders. Some individuals affected with each of these dominant disorders show typical levels of social responsiveness, while others show a social profile indistinguishable from idiopathic ASDs. The distribution of autism traits, as measured by the SRS, appears to be normal with a shifted mean for CFC and CS. For NF1 and NS, the distributions appear to have an increased variance but are not strictly bimodal. This could reflect underlying RASopathy mutational (and thus biochemical) heterogeneity, or could be influenced by other factors. Notably, the CS subjects had the lowest variance in SRS scores when compared to other RASopathies, and they also have the most homogeneity in primary CS mutation, with two neighbouring amino acid substitutions affecting the same protein accounting for nearly all known mutations (over 75% of subjects). An important future direction will be to assess whether specific mutation-phenotype relationships can explain any of the within-disorder variance.
Interestingly, although RASopathies affect males and females equally, autism traits in RASopathy probands (but not unaffected siblings) were more evident in males. This, along with possible sibling correlation (below), may suggest that a RASopathy mutation is strongly sensitising, but autism traits are otherwise determined similarly to the general population where males show much higher prevalence of autism compared with females.
Subjects with NF1 and CFC (and more modestly, CS) showed correlation in social responsiveness with RASopathy-unaffected siblings at a level consistent with the general population, although these correlations were not highly significant and could be influenced by outliers. Correlation could suggest that apart from a main effect of the RASopathy mutation, background genetic variation may be important in variability within disorder. An alternative explanation is that the parent raters who scored both siblings show a similar bias in evaluating both children, although the difference between mean scores in affected and unaffected siblings was highly significant. Previous research in autism families shows that, in fact, the opposite is often true, that parents show a contrast bias in differentiating their children which could minimise sibling correlation.72
However, we do not see evidence for sibling correlation in NS when all probands are included, driven by the probands scoring above average for their RASopathy group. This, in conjunction with lack of sex-bias, potentially indicates a truly epistatic mechanism determining severe social cognitive impairment within NS and generating bimodality. An alternative explanation is that the clinical syndrome in this case is not the appropriate classification if different genetic mechanisms (eg, upstream vs downstream mutations) have vastly different main effects that we have not accounted for since we lack mutation information on a large proportion of our sample.
Upon clinical evaluation, four out of six subjects with high scores on parent questionnaires scored in the ASD range on the gold standard autism diagnostic observation, although clinical impression was not always of ‘typical’ autism. By contrast, we found the larger population of RASopathy subjects and idiopathic ASD subjects who scored above threshold on the SCQ showed indistinguishable subscale profiles on the SRS by PCA, suggesting that the high RASopathy scores were not driven by unequal domains of impairment or other factors, such as cognitive or attentional impairment. This analysis also suggested that there is not significantly more symptom homogeneity in RASopathies compared with idiopathic heterogeneous ASDs. Future study including additional measurements characterising a developmental profile of change over time would be useful. We found in this small sample that SRS score in the ‘severe’ range and SCQ>15 were good predictors of gold-standard instrument in-person research criteria.
This systematic study offers empirical support that autism traits are associated with developmental Ras/MAPK pathway dysregulation. It suggests that individuals affected by RASopathies should be evaluated for social communication challenges and offered treatment in these areas. This is the first strong evidence that multiple members of a well-defined biochemical pathway can contribute to autism traits. Future studies could explore potential modifying or epistatic factors contributing to variation within the RASopathies and the role of Ras/MAPK activation in idiopathic ASDs.
Acknowledgments
We are grateful to Kim Taylor, Bryna Siegel, Lindsey Criswell, Jane Gitschier, Carol Mathews, Ophir Klein, and Anthony Wynshaw-Boris for helpful discussion, to Jae Eun Choi for her assistance in diagnostic testing, and to Keren Messing-Guy, Julie Lustig, and Allison Kiang for their help in subject recruitment and data entry. We also thank NF, Inc., Children's Tumor Foundation, Noonan Foundation, CFC International, Costello Syndrome Family Support Network, and Costello Kids for their contribution to our recruitment efforts.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
-
Contributors BA: data acquisition, data analysis, manuscript drafting and revising. ILC: data acquisition, data analysis, manuscript drafting. FW: data acquisition, data analysis, data interpretation. GD: data analysis and interpretation. NE and TR: data acquisition. SR: data interpretation. EJM: data interpretation, revising for intellectual content. RLH and KAR: data acquisition, data interpretation, revising for intellectual content. CEB: data acquisition, revising for intellectual content. LAW: study conception and design, data acquisition, data analysis, data interpretation, manuscript drafting and revising, final approval of version published.
-
Funding The work was supported by the NIH New Innovator 1DP2OD007449 (LAW), SFARI Explorer #257754 (LAW), an IHG Exploratory Grant (LAW), a Staglin Family/IMHRO Assistant Professorship (LAW), NIMH R34 MH089299-01 (CEB) and the Carol Moss Spivak Foundation (CEB).
-
Competing interests LAW has received an honorarium from the Simons Foundation for the SFARIgene.org website, and the Simons Foundation has funded one of the research grants supporting part of this study.
-
Ethics approval UCSF Committee on Human Research.
-
Provenance and peer review Not commissioned; externally peer reviewed.