Article Text

Abstract
Background Consensus clinical diagnostic criteria for neurofibromatosis type I (NF1) include café-au-lait macules and skinfold freckling. The former are frequently the earliest manifestation of NF1, and as such are of particular significance when assessing young children at risk of the condition. A phenotype of predominantly spinal neurofibromatosis has been identified in a small minority of families with NF1, often in association with a relative or absolute lack of cutaneous manifestations. An association with splicing and missense mutations has previously been reported for spinal neurofibromatosis, but on the basis of molecular results in only a few families.
Method Patients with spinal NF1 were identified through the Manchester nationally commissioned service for complex NF1.
Results Five families with spinal NF1 were identified, with a broad spectrum of NF1 mutations, providing further evidence that this phenotype may arise in association with any genre of mutation in this gene. Pigmentary manifestations were absent or very mild in affected individuals. Several further affected individuals, some with extensive spinal root tumours, were ascertained when additional family members were assessed.
Conclusions Clinical NF1 consensus criteria cannot be used to exclude the diagnosis of spinal NF1, especially in childhood. This emphasises the importance of molecular confirmation in individuals and families with atypical presentations of NF1.
- Clinical genetics
- Dermatology
- Genetics
- Other neurology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Neurofibromatosis 1 (NF1) is an autosomal dominant condition caused by mutations in the NF1 gene on chromosome 17.1 Considerable inter- and intra-familial variability can complicate clinical ascertainment of affected individuals.2 Consensus criteria for the clinical diagnosis of NF1 are well established,3 but these may not be effective in very young children, especially where there is no family history of the disorder. Additional criteria such as T2 hyperintensities on MRI of the brain in childhood4 have been suggested, but these too are not universally present. Notwithstanding these limitations, a definitive clinical diagnosis can be made for a large proportion of patients with NF1. In contrast, processes for molecular confirmation of NF1 have been complicated by the very large size of the gene, its homology with many pseudogenes, and a lack of mutational hotspots. Very limited evidence for genotype–phenotype correlation exists, in keeping with the likelihood of haploinsufficiency as the molecular mechanism causing pathogenicity for the majority of mutations identified to date. Exceptions to this include the relatively distinct phenotype of patients with whole gene deletions of NF1,5 where the altered genomic landscape may play a role, and the very mild café au lait (CAL) only pattern in patients with a common three base pair in frame deletion.6 More recent evidence of association of specific groups of mutations with optic pathway glioma7 and pulmonary stenosis8 has also emerged.
Situations in which molecular confirmation is absolutely necessary, such as for prenatal or pre-implantation genetic diagnosis, have to date been relatively rarely encountered in NF1. However, the recognition of disorders with overlapping clinical or radiological phenotypes but differing prognoses, including schwannomatosis9 ,10 and Legius syndrome,11 ,12 emphasise the importance of molecular diagnosis in an increasing spectrum of patients with NF1 and related disorders.
Within the NF1 population, various subtypes of disease have now emerged, including groups of families where several individuals have a high load of spinal tumours (‘spinal NF1’). As is the case for most patients with NF1, these families have generally been found to have private mutations, but an association between the spinal phenotype and splice and missense variants has previously been suggested.13 Five further families are described here in which affected individuals have a high spinal tumour load, due to a range of mutations in NF1. Minimal pigmentary manifestations were present in affected people, complicating clinical diagnosis and making it very difficult to reassure at-risk individuals in these pedigrees. This circumstance emphasises the value of molecular testing for definitive identification of individuals at high risk of problematic neurofibromas who will require early investigation of any spinal symptoms in particular, and appropriate MRI surveillance. Clinical presentations, molecular analysis, and the implications of these findings for families with absent or non-diagnostic pigmentary features of NF1 are discussed.
Methods
Affected individuals were ascertained through the Manchester nationally commissioned service for complex NF1 and examined (by DGRE, SMH or OQ) in its specialist clinics (based in Manchester). Where indicated, MRI scans of the neuroaxis and/or whole body were carried out, and interpreted along with the clinical phenotypes in the context of a multidisciplinary team discussion. Permission for inclusion of clinical and imaging details was sought from the patients reported here.
Molecular analyses were carried out in the clinical pathology accredited Manchester Regional Genetics Laboratory and in the Genetics Research and Development laboratory in Cardiff. RNA and genomic DNA were prepared from peripheral blood samples. RNA was reverse transcribed to cDNA using standard procedures, and direct sequencing performed to demonstrate splicing abnormalities or mutations within the coding sequence. Mutation status was confirmed in genomic DNA. Multiplex ligation dependent probe amplification (MLPA) for dosage analysis was additionally performed in samples without a clearly pathogenic mutation; for example, where a novel sequence variant with uncertain pathogenicity had been identified.
Family 1
This family had individuals affected across at least four generations, as shown in figure 1A. The proband, IV:5, presented aged 8 years in New Zealand, when a neurofibroma had been excised from his right arm. At the age of 20 years he developed difficulty in walking, pain in his right leg, and occasional paraesthesia in his hands. Peripheral subcutaneous neurofibromas, some of which were painful, a thoracolumbar scoliosis, and a neurological deficit in the lower limbs were identified on examination. Serial MRI showed progression of multiple peripheral nerve neurofibromas in his legs and bilaterally in his cervical spine. From the age of 21 he underwent excision of multiple symptomatic cervical spine neurofibromas. At 23, multiple subcutaneous lesions were excised from his right thigh, and were confirmed as benign neurofibromas with plexiform elements. Skin examination at age 25 confirmed just one CAL macule, six subcutaneous neurofibromas, and one cutaneous neurofibroma. Lisch nodules were noted, but neither axillary nor inguinal freckling were present.

(A–E) Pedigrees of families 1–5.
IV:5's mother, III:5, was examined in the genetics department at the age of 50 years. She had no CAL, and skinfold changes were also absent apart from a few unilateral axillary freckles. Multiple painful subcutaneous neurofibromas, a plexiform neurofibroma, spinal neurofibromas, scoliosis, and Lisch nodules were all present.
III:2 was diagnosed with NF1 in her third decade, when a lump excised from her mouth was found to be a neurofibroma. In each of her three pregnancies (at 26–30 years of age), she developed new cutaneous neurofibromas. She was first reviewed in the genetics department at the age of 52, and painful cutaneous and subcutaneous neurofibromas were noted which were subsequently histologically confirmed. She had no skin pigmentary changes nor any symptomatic spinal tumours, and therefore MRI imaging was not performed at that time.
III:2's three sons, IV:2, IV:3, and IV:4, were each examined on account of their 50% risk of NF1. Two each had one CAL documented in childhood and one additionally had three subcutaneous nodules which subsequently disappeared. No other pigmentary changes, including Lisch nodules, were identified in any of these individuals. They have remained well with no signs of NF1, but this diagnosis could not be confidently excluded clinically until they were well into adulthood, given the minimal pigmentary findings of the condition in other family members, particularly their mother.
III:1 had a longstanding diagnosis of NF1 on the basis of subcutaneous neurofibromas, several of which had been removed and histologically analysed; his daughter (IV:1) also had CAL from early life. He developed deafness and had a brain scan at the age of 54 years. No cause for deafness was identified, but an asymptomatic cervical spinal neurofibroma with an intradural component was seen, with bilateral impingement on the C2 nerve roots. This required surgery, due to the risk of spinal cord compression. On examination at the age of 62, over 100 cutaneous and subcutaneous neurofibromas were present, as were axillary freckles and Lisch nodules. In contrast, only seven CAL (three of which required ultraviolet light for visualisation) were identified.
III:1's daughter, IV:1, was diagnosed in early childhood with NF1 on account of her family history and presence of CAL. At the age of 7 years, she had no learning difficulties and only four or five CAL patches, but she also had depigmented areas of skin over her back. By 22 years of age, she had six CAL, minimal axillary and inguinal freckling, and a small number of histologically proven neurofibromas.
II:2, III:5's late father, was reported as having a severe disfigurement due to skin lumps, and had died from what was highly likely to have been a malignant peripheral nerve sheath tumour (MPNST). His mother (I:1) was also thought by the family to have been affected with NF1, as her skin was covered in lumps. It was also reported that his late father (I:2, a first cousin of I:1) could have been similarly affected.
For all living affected individuals in this family, a diagnosis of NF1 could be made by application of the clinical consensus criteria. However, this was only possible for many of them well into adulthood, due to both the paucity of pigmentary manifestations, and the late onset of visible or symptomatic tumours in most individuals. What was not clinically straightforward was to exclude NF1 in at-risk individuals. This was a major source of anxiety for several family members, who were worried about risks to their offspring of this disabling condition. Pre-implantation genetic diagnosis was considered by IV:5 and his wife due to the burdensome nature of his own and his mother's (III:5) condition, but was not available when he and his wife were starting a family. After extensive consideration within the family, his two daughters were each tested at birth for the recently identified familial mutation, c.6364+2T>G, which had been found to result in exclusion of exon 33 and a subsequent frameshift resulting in a prematurely terminated protein. They were found to be unaffected.
Family 2
The proband, I:1 (figure 1B), was seen in the genetics clinic at the age of 30 years after identification of multiple nerve root tumours. She had suffered back pain, predominantly in the cervical and lumbar regions, since puberty, which worsened in each of her three pregnancies and was causing significant disability. She was under follow-up in the pain clinic which had prescribed gabapentin, amitriptyline, and tramadol. MRI scans (figure 2A,B) demonstrated mild thoracic scoliosis and enlargement of multiple segmental nerve roots from C5 to T1 and from L2 to S1. Degenerative disc change was also noted at L4/L5. Vestibular schwannomas were absent. One deep neurofibroma was present in the left thigh, and one subcutaneous neurofibroma on the right shoulder. Her height was 173 cm (91st–98th centile) and her head circumference was 56.8 cm (75th–91st centile). No CAL, freckles or plaque skin change (the last as seen in NF2) were present. Three to four Lisch nodules were seen in the right eye, and one in the left. The differential diagnosis on clinical and radiological grounds rested between schwannomatosis and NF1. A frameshift NF1 mutation, c.5993dupC, resulting in a premature termination codon 10 residues downstream, p.(Thr1999Asnfs*10), was identified in her lymphocyte DNA.

{kind=link}
{kind=link}
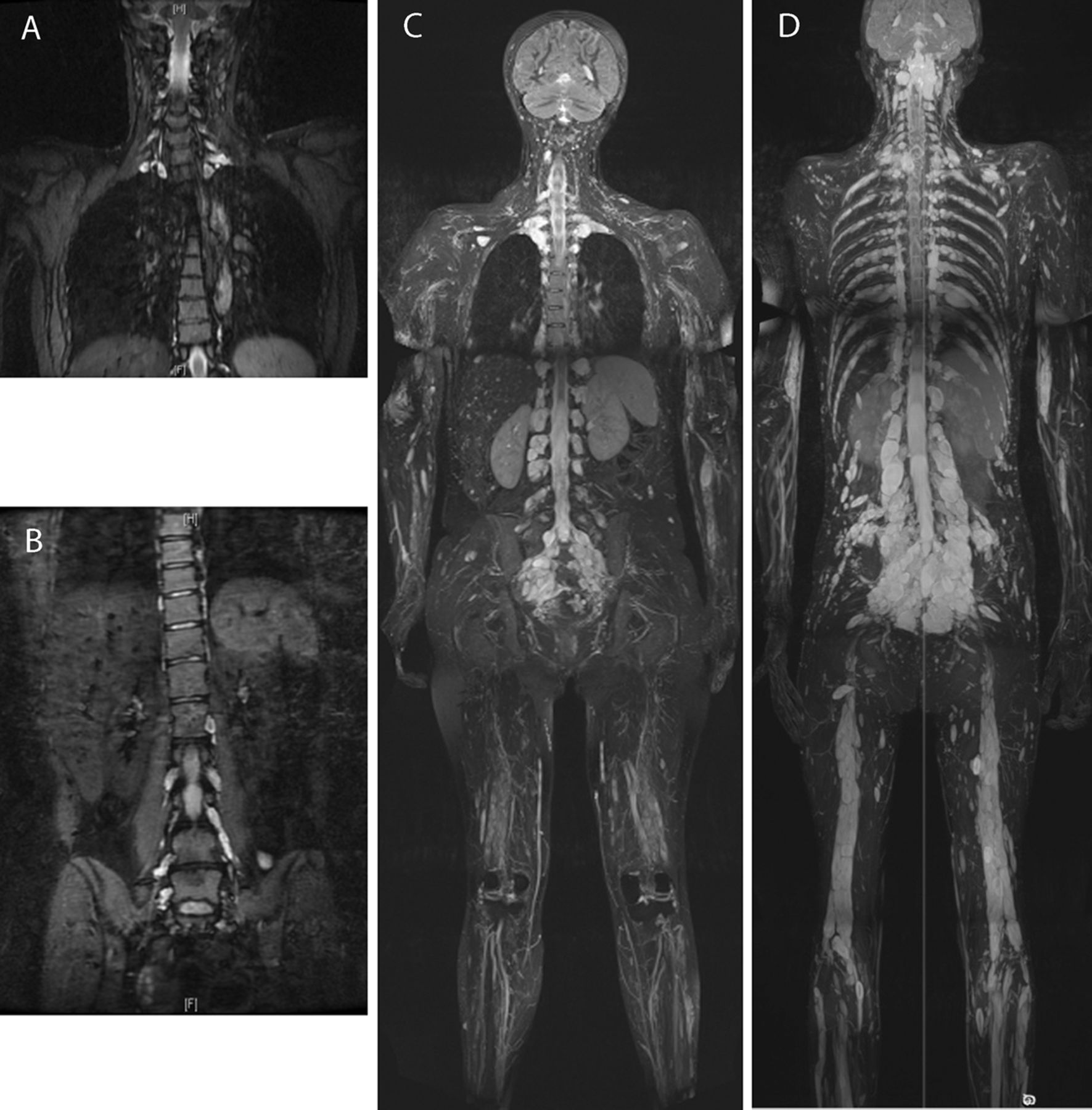
(A and B) Coronal short TI inversion recovery (STIR) images of (A) cervical and (B) lumbosacral spine of patient I:1 of family 2. At 30 years of age, multiple lesions are present bilaterally, most marked in the mid and lower cervical levels (A; arrowed) and in the sacrum, particularly of the S1 nerve root (B; arrowed). A mild thoracic kyphoscoliosis is also seen (A), and degenerative change of the L5/S1 intervertebral disc (B). (C) Coronal maximum intensity projection (MIP) 2 cm thick STIR image from the whole body MRI scan of patient II:1 of family 5. At 43 years of age, spinal nerve sheath tumours can be seen bilaterally at nearly every vertebral level: cervical and lumbar lesions are demonstrated. Alongside the extensive nerve root tumours, multiple subcutaneous lesions can be seen in the scalp (arrow) and extremities. (D) Coronal curved plane reformat MIP STIR image from the whole body MRI scan of patient III:1 of family 5. At 20 years of age, similar to his mother's presentation, spinal nerve sheath tumours can be seen bilaterally at nearly every vertebral level. A particularly large lesion, approximately 30 mm in smallest diameter, is seen in the sub-occipital area.
I:1's three children were each assessed in clinic regarding the likelihood of being affected with NF1. II:1 and II:3 each had features of attention deficit hyperactivity disorder (ADHD) and learning delay (at the ages of 8 and 5 years, respectively), with II:3 also having a history of hypotonia and coordination difficulties. All head circumferences were around the 50th centile. II:2 (aged 6) had one CAL, but no other pigmentary features or abnormalities were present in any of the children. After appropriate counselling, genetic testing was performed which demonstrated II:2 and II:3 each carried the mutation, indicating their need for long-term follow up for spinal sequelae.
Family 3
The proband, II:2 (figure 1C), was referred to the clinic at the age of 32 years following the identification of multiple symptomatic tumours on the lumbar spinal nerves, and a plexiform tumour in the right flank. MRI scanning had been undertaken because of pain and paraesthesia in his right leg, for which he had been prescribed tramadol and pregabalin. Positron emission tomography showed low activity in the lesions, making it unlikely that the observed tumours were malignant, and their appearance was unchanged on a repeat MRI scan 1 year later. Of note, his younger brother (II:3) had died at age 26 of glioma, and his older maternal half-brother (II:1) had been diagnosed with bladder cancer aged 43. His parents (I:1 and I:2) were healthy, with no features of NF1, and there was no other family history of malignancy of other tumours. II:2 had no cutaneous manifestations of NF1: no CAL macules, freckling or dermal neurofibromas were present. Genetic testing of NF1 and SMARCB1 was initiated, to clarify whether the tumours identified radiologically were neurofibromas or schwannomas. A novel variant, c.7083C>T, encoding p.(Phe2361Leu), was identified in NF1. No abnormalities were identified in sequencing of SMARCB1, or by MLPA of either gene. Analysis of DNA from other family members to assess segregation of the NF1 variant was not possible.
Family 4
The proband, II:1 (figure 1D), was found to have multiple cervical nerve sheath tumours on CT scan, which was performed following a head injury at the age of 25 years. When assessed, he reported numbness and paraesthesia of his right arm, and numbness of his right leg since childhood, but with no recent progression of these symptoms. He was also found to have some small subcutaneous nodules over his neck and scalp. The largest lesion seen on the scan was a right-sided C6/C7 nerve sheath tumour, compressing the cord but without signal change. In view of his symptoms, he was started on pregabalin 75 mg twice daily. He had no pigmentary features of NF1, hence the differential diagnosis was evenly poised between schwannomatosis and spinal NF1, and genetic testing was performed. A pathogenic splice site mutation in NF1, c.2002-3C>G, was identified. In the light of this, further asymptomatic family members were evaluated.
In II:1's father, I:1, no CAL marks or other cutaneous stigmata of NF1 were present, but MRI scanning of his spine demonstrated multiple lesions of the lumbar nerve roots, most prominently a mass at the right side of L5 with secondary lumbar canal stenosis at L4/5. Foraminal and extraforaminal portions of other lumbar nerve roots also appeared thickened.
On clinical examination, II:1's sister, II:2, was found to have several CAL patches on her back and neck, and some axillary freckling, but no neurofibromata. Results of MRI scanning of her spine were awaited. A further sibling was also assessed, but was not available to ask for permission for publication of clinical details. I:1's sister, I:2, was understandably anxious about the risk of NF1 to herself and her children. She opted for predictive genetic testing, and it was demonstrated that she did not carry the familial mutation.
Family 5
The proband, II:1 (figure 1E), developed multiple subcutaneous nodules from her teenage years. At the age of 26 years, she had a large lesion, reportedly a neurofibroma, excised from the right posterior aspect of her neck. She subsequently had excisions of several other subcutaneous lesions from her back, but remained generally well until the age of 31, when she presented with increasing right leg and groin pain. Spinal MRI showed tumours present on nerve roots throughout the neuroaxis, particularly around the cauda equina, and a large retroperitoneal mass. Surgical intervention was not indicated for any of these, and she remained under surveillance with annual serial MRI scans. A possible diagnosis of neurofibromatosis type II (NF2) was raised when a possible lesion at the CPA was identified at the age of 39, which was associated with right-sided vestibular symptoms and tinnitus. However, pure tone audiometry was normal, no CPA abnormality was seen on interval MRI, and her symptoms did not progress. At the age of 44, she was reviewed in the genetic clinic. She had a very extensive burden of nerve sheath tumours, involving the large majority of spinal nerve roots (figure 2C), but no CAL patches or Lisch nodules. Freckling was present across the upper trunk and in the axillae. The absence of CPA abnormality indicated that the diagnosis of NF2 could be confidently excluded, and the previous clinical impression of spinal NF1 was confirmed. Sequencing of lymphocyte DNA demonstrated a previously reported pathogenic mutation in NF1, c.2543G>A, predicting p.(Gly848Glu). Her body habitus was typical for NF1, with short stature of 146 cm (<0.4th centile), some facial features suggestive of Noonan syndrome, and a relatively large head circumference (59 cm, >97th centile).
Her mother, I:1, had a subcutaneous lump, histologically most consistent with a plexiform neurofibroma, removed from her back at the age of 62. She had no pigmentary changes in her skin nor Lisch nodules, but carried her daughter's mutation in lymphocyte DNA. I:1's mother had also had a lump removed, and died at a young age, but this history could not be further elucidated.
II:1's son, III:1, was reviewed at the age of 20, in light of his 50% risk of spinal neurofibromatosis. He was asymptomatic and of normal stature (168 cm), with a normal head circumference of 57.5 cm, and had no CAL patches, axillary freckling or subcutaneous neurofibromas. No Lisch nodules were present, but he did have some facial features consistent with NF1/Noonan syndrome and he also had pectus carinatum. He had discrete lesions of approximately 1 cm in diameter palpable in the left anterior triangle of the neck. He was tested within a predictive testing protocol for the NF1 mutation identified in his mother, and found to carry this. Whole body MRI (figure 2D) showed an extensive tumour burden, including a particularly large lobulated lesion in the occipital/posterior neck region (figure 2D). Despite his slim build, this was not palpable. An arachnoid cyst of the left middle cranial fossa was also demonstrated.
Results
Clinical features of affected individuals are summarised in table 1, and the mutations identified in each family are shown in table 2.
Clinical features in affected individuals
NF1 mutations identified in the five families
Discussion
Spinal tumours are common in NF1, but appear to be only rarely symptomatic. However, there is a relative paucity of clinical literature regarding this. Thakkar et al16 identified such tumours in 40% of 30 asymptomatic patients with NF1, but only 2% of people in their large cohort (n=1400, aged 5–56 years) had developed symptomatic spinal tumours, and long term clinical follow-up data of such lesions are lacking. Individuals with symptomatic spinal tumours and typical cutaneous features of NF1 do not appear more likely than unselected NF1 patients to have a family history of the disorder. Conversely, multigenerational familial clustering of the spinal NF1 phenotype with few cutaneous features is well recognised,17 suggesting that individuals carrying mutations causing this phenotype may be more likely than the NF1 population in general to have children. Missense mutations have been highly overrepresented in the variants reported in association with spinal NF1 (nine of 15 mutations included in the human genome mutation database18), and a significant further proportion have been substitutions affecting splicing.13 ,19 ,20 It has therefore been postulated that the different clinical phenotype observed in these families could be a result of milder molecular effects of these mutations,17 but tissue specific effects may also be of importance. In support of the latter hypothesis, a rare familial phenotype in which affected individuals had optic nerve glioma, other central nervous system (CNS) tumour, or both,21 has been reported in association with a splice donor site mutation in NF1, resulting in skipping of exon 29. At least two individuals with CNS tumours who carried this mutation did not have typical skin manifestations of NF1, but others did. Differential tissue specific effects for splicing of exon 29 of NF1 have also been suggested by the work of Park et al,22 with expression of certain transcripts including this exon being limited to brain, of the tissues examined. This phenomenon could be a key contributing factor to this family's unusual phenotype. Such an example further highlights the importance of mutational analysis, particularly at the RNA level, in families with atypical NF1 phenotypes. The data from our series broadly support the hypothesis that splice and missense variants are the main causes of a spinal limited NF1 phenotype (as observed in four of five families described here), but that frameshift mutations may also, rarely, result in this phenotype, as has previously been reported.23 Similarly, Pizzuti et al24 reported a multi-exon deletion, resulting in nonsense mediated decay of the transcript, in a family with spinal neurofibromatosis where a 73-year-old man was only found to be affected, with typical spinal MRI findings, when his daughter was diagnosed.
The late onset of symptomatic manifestations of spinal NF1 has several implications for clinical practice. Tumour risk, either by malignant transformation of neurofibromas, or due to development of other neoplasia, has not yet been satisfactorily established in the small cohort of patients diagnosed with familial spinal NF1, though internal tumour burden is a well recognised risk factor for development of MPNST.25 Late onset of the NF1 phenotype may be an important contributor to the difficulty in establishing such risks. The large burden of morbidity identified to date in this patient group is due to the effects of non-malignant spinal tumours, with high rates of pain, reduced mobility and paraparesis. Patients with NF1 and a heavy internal burden with spinal root involvement represent a considerable management problem. In the future it is likely that they will be treated with pathogenesis based medical therapies that are now being shown to be effective in mouse models.26
Many of the affected patients reported here endured several years of such symptoms before the diagnosis was suspected. As the first affected individual in a family, this may be unavoidable if no further stigmata of NF1 are evident. Increasing professional awareness of the possibility of the diagnosis in patients with subtle or few other manifestations of NF1 is important in order to optimise management, as neurosurgical outcomes may be improved by presymptomatic intervention in affected individuals. Of note, due to the predominantly extra-axial location of the tumours, an extensive tumour burden may be present in asymptomatic patients, and appropriate counselling regarding this should be undertaken before MRI scanning of these individuals. The frequently late onset of symptoms also means that affected individuals may have children of their own before their genetic risk becomes apparent. Predictive genetic testing protocols may therefore be important for asymptomatic at-risk individuals in these families, as the usual clinical assessment in childhood cannot be relied upon to confirm or refute the diagnosis. Diagnostic testing of symptomatic individuals is important for molecular confirmation, and may obviate the need for surgical biopsy for histological confirmation of the diagnosis, again benefiting patient care.
Conclusion
The spinal phenotype of NF1, without associated pigmentary manifestations, can occur in association with a range of mutations in NF1. Molecular testing of the gene is warranted in patients with atypical presentations such as possible spinal NF1, particularly as this diagnosis may not be clinically or radiologically distinguishable from related conditions such as schwannomatosis.
Acknowledgments
DGRE is an NIHR senior investigator. This work was facilitated by the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network.
References
Footnotes
-
Contributors EMMBW, DGRE, SS and SH wrote the body of the paper. DGRE, SH, EMMBW, SS, OQ and TC generated the clinical data. ES and MU were responsible for the molecular testing of NF1, and RWW performed and interpreted the imaging studies. All authors have reviewed and edited this manuscript.
-
Funding EMMBW is a Wellcome Trust funded Clinical Research Training Fellow (090120).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This is a retrospective case series. All of the data included was obtained in the course of clinical care.
-
Provenance and peer review Not commissioned; externally peer reviewed.