Article Text

Abstract
Background Kaufman oculocerebrofacial syndrome (KOS) is a developmental disorder characterised by reduced growth, microcephaly, ocular anomalies (microcornea, strabismus, myopia, and pale optic disk), distinctive facial features (narrow palpebral fissures, telecanthus, sparse and laterally broad eyebrows, preauricular tags, and micrognathia), mental retardation, and generalised hypotonia. KOS is a rare, possibly underestimated condition, with fewer than 10 cases reported to date. Here we investigate the molecular cause underlying KOS.
Methods An exome sequencing approach was used on a single affected individual of an Italian consanguineous family coupled with mutation scanning using Sanger sequencing on a second unrelated subject with clinical features fitting the disorder.
Results Exome sequencing was able to identify homozygosity for a novel truncating mutation (c.556C>T, p.Arg186stop) in UBE3B, which encodes a widely expressed HECT (homologous to the E6-AP carboxyl terminus) domain E3 ubiquitin-protein ligase. Homozygosity for a different nonsense lesion affecting the gene (c.1166G>A, p.Trp389stop) was documented in the second affected subject, supporting the recessive mode of inheritance of the disorder. Mutation scanning of the entire UBE3B coding sequence on a selected cohort of subjects with features overlapping, in part, those recurring in KOS did not reveal disease-causing mutations, suggesting phenotypic homogeneity of UBE3B lesions.
Discussion Our data provide evidence that KOS is caused by UBE3B loss of function, and further demonstrate the impact of misregulation of protein ubiquitination on development and growth. The available clinical records, including those referring to four UBE3B mutation-positive subjects recently described as belonging to a previously unreported entity, which fits KOS, document the clinical homogeneity of this disorder.
- Clinical genetics
- Developmental
- Diagnosis
- Genome-wide
- Molecular genetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Kaufman oculocerebrofacial syndrome (KOS) (MIM 244450) is the eponym for the disorder originally described by Kaufman, Rimoin and co-workers 40 years ago in four of seven sibs born to unaffected parents exhibiting mental retardation, microcephaly, upward palpebral fissures, microcornea, severe myopia, optic atrophy, preauricular skin tags, high arched palate, and micrognathia.1 KOS is an extremely rare, possibly underestimated condition, with fewer than 10 affected individuals reported to date.2–4 Based on the available records, main features include microbrachycephaly, distinctive face, eye anomalies (small cornea, strabismus, myopia, and pale optic disk), mild to severe psychomotor retardation, hypotonia, delayed growth, and skeletal features (bell shaped thorax with telethelia, long and thin fingers). The recognisable facial appearance is characterised by sparse and laterally broad eyebrows, upslanting and narrow palpebral fissures, telecanthus, anteverted nostrils with columella below alae nasi, and micrognathia. Small, poorly formed and frail teeth and a high, narrow palate have commonly been reported.
A recurrent feature of KOS is the presence of anomalies of structures that are derived from the first and second pharyngeal arches, since external ear anomalies (cup shaped ears and stenotic auditory canal) and preauricular tags have been described to occur concomitantly with micrognathia in affected subjects. The presence of blepharophimosis and mental retardation, with or without microcephaly and micrognathia, is described in a heterogeneous group of multiple congenital malformation disorders, including Dubowitz syndrome (MIM 223370), Marden–Walker syndrome (MIM 248700), Smith–Lemli–Opitz syndrome (MIM 270400), and Ohdo syndrome (MIM 249620) and related phenotypes.5 In those conditions, however, individual combinations of congenital anomalies, facial gestalt, and natural history are definitely different from the constellation of features occurring in KOS. Here, an exome sequencing approach was employed to identify UBE3B (MIM 608047), which encodes an E3 ubiquitin-protein ligase, as the gene mutated in KOS, confirming this disorder as a distinct nosologic entity inherited as an autosomal recessive trait.
Subjects and methods
Patients and families
Two unrelated individuals with clinical features fitting KOS were included in the study (figure 1A, see online supplementary table S1). In the subjects, karyotype and subtelomeric fluorescence in situ hybridisation (FISH) analyses were normal. Similarly, high resolution array comparative genomic hybridisation analysis did not disclose any disease-causing genomic rearrangement in either proband. Twelve subjects who had no confirmed clinical diagnosis and sharing features overlapping, in part, those recurring in KOS, together with the patient originally reported by Briscioli et al,6 were all included for mutation scanning of the UBE3B gene (see online supplementary table S2). For all patients, clinical data and biological material collection and storage were obtained from the participating families after they provided written informed consent.
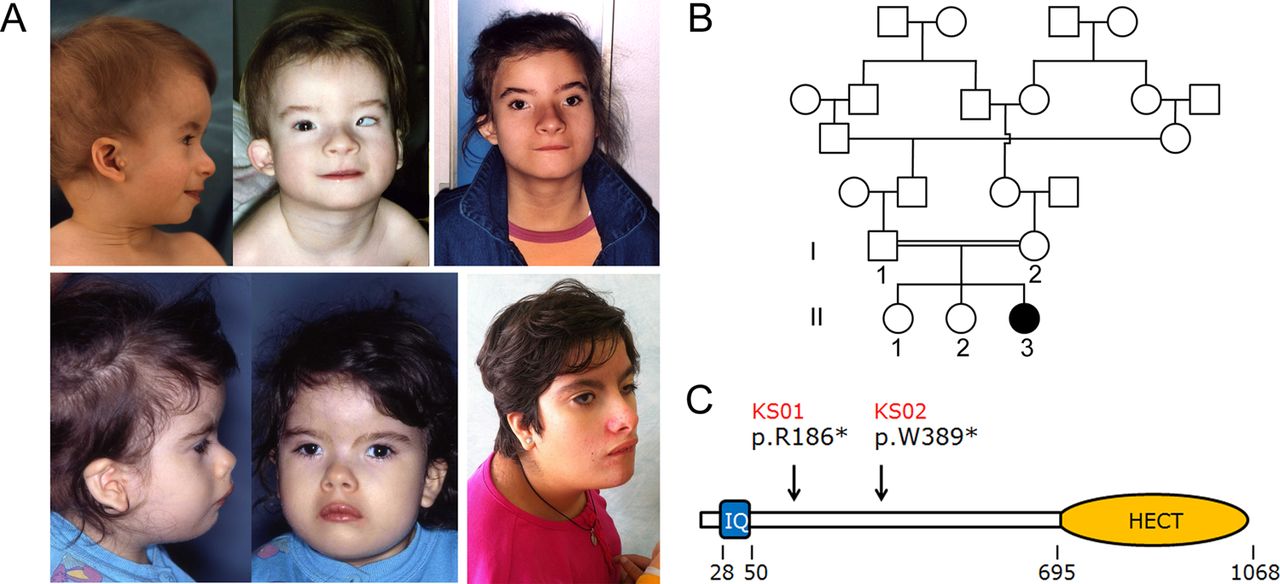
Craniofacial features in Kaufman oculocerebrofacial syndrome and UBE3B domain structure. (A) The recognisable facial gestalt of the two affected subjects (UCSC_KS01, above; UCSC_KS02, below). Note the distinctive facies of the two affected subjects with microcephaly, sparse eyebrows with unusual profile, upslanting and narrow palpebral fissures, strabismus, cup shaped ears, preauricular tags, and micrognathia. (B) Pedigree structure of family UCSC_KS01. Genotyped members are indicated. (C) Scheme of the UBE3B domain structure. The ubiquitin ligase comprises an N-terminal IQ domain (IQ), and a C-terminal catalytic domain (homologous to the E6-AP carboxyl terminus). Numbers below the diagram indicate the amino acid boundaries of those domains. The location of identified mutations in patients UCSC_KS01 and UCSC_KS02 is also reported.
Exome sequencing and sequence data analysis
Targeted enrichment and massively parallel sequencing were performed on genomic DNA extracted from circulating leucocytes of patient UCSC-KS01. Exome capture was performed using NimbleGen SeqCap EZ Exome V. 2.0 (Roche), and sequencing using a HiSeq2000 instrument (Illumina). Paired-end reads were aligned to human genome (UCSC GRCh37/hg19) with the Burrows–Wheeler Aligner (BWA V. 0.5.9-rc1).7 Presumed PCR duplicates were discarded with the Picard's MarkDuplicates utility (http://picard.sourceforge.net). Local realignment and base-quality-score recalibration were performed with the Genome Analysis Toolkit (GATK).8 Single nucleotide polymorphisms (SNPs) and indels were identified with the GATK Unified Genotyper,9 excluding from further analyses variants which failed to meet any of the following criteria: alignment quality >50; quality-by-depth score >1.5; variant resulting from >3 reads having unambiguous mapping (the number being >1/10 of all aligned reads). Variants were filtered against available public (1000 Genomes Project and dbSNP135) and in-house databases. Functional annotation of variants was performed by using snpEff V. 2.0.5d.10 Variants within segmental duplications and variants outside highly conserved regions in vertebrates were filtered using wAnnovar (http://wannovar.usc.edu).
Variant validation and mutation analysis
Sequence validation and segregation analyses for all the candidate variants as well as mutation scanning of the entire UBE3B coding sequence (exons 1–26) (NM_130466) was performed by Sanger sequencing using an ABI Prism 3500 Genetic Analyzers (Applied Biosystems) and the ABI BigDye Terminator Sequencing Kit V.3.1 (Applied Biosystems). Primer pairs designed to amplify the UBE3B coding exons and their intron boundaries are listed in online supplementary table S3. Sequence comparisons and analyses were performed using the Sequencing Analysis Software V.5.4 (Applied Biosystems).
Results
Exome sequencing obtained of 52 million 90 bp paired-end reads. Following the alignment pipeline, target region coverage was 99.2%, with average sequencing depth on target of 56×. The total number of variants called was 33 657 (32 111 SNPs and 1546 small INDELS (insertions and deletions)). Among them, 32 012 are annotated in dbSNP135. The remaining 1645 variants were filtered to retain 323 sequence changes located in exons and splice sites with any functional effect, which were selected for further analyses. Variants annotated in the 1000 Genomes Project variant and in-house databases were also removed, leading to a total of 152 previously unreported variants.
Since there was evidence of consanguinity in the family (figure 1B), we considered homozygosity for a private variant as the most likely event underlying the trait. Autosomal recessive pattern of inheritance was also consistent with the multiple occurrences in sibs of both sexes with unaffected parents in the originally reported family.1 According to a recessive model, a list of 11 variants in eight candidate genes was compiled. Based on the hypothesis of homozygosity by descent, the list of candidates was reduced to five genes (PLD1, NDUFB9, PTCHD3, UBE3B, and VWA3B). Given the relevance of ubiquitination mediated processes in development and morphogenesis, and the dramatic functional impact of the sequence change (c.556C>T, p.Arg186stop), we considered UBE3B as the most promising disease gene candidate. UBE3B encodes a member of the ‘homologous to E6-AP carboxyl terminus’ domain containing E3 ubiquitin-protein ligase enzyme family.11 The protein is characterised by an IQ domain near to the N-terminus (residues 28 to 50), which mediates protein binding, and a HETC domain at the C-terminus (residues 695–1068) (http://smart.embl-heidelberg.de/) (figure 1C). As the latter represents the catalytic site, interacting with the E2 conjugase and mediating ubiquitin transfer to the protein substrate, the nonsense mutation was predicted to result in a catalytically impaired protein.
Sanger sequencing of the relevant coding exon confirmed the homozygous condition for the c.556C>T nucleotide substitution in the affected subject (figure 2A), and excluded homozygosity for the lesion in the two unaffected sibs. Occurrence of the nucleotide change was also excluded in 400 population matched control subjects. Consistent with the view that this nonsense change in UBE3B was the causative mutation, the other two of the five unannotated homozygous variants confirmed by Sanger sequencing were documented to occur as homozygous changes also in one of the two unaffected sibs, excluding their causal link with the disease. Similarly, segregation analysis of validated variant pairs identified in the BRD8, IQGAP2, and SYNE2 genes excluded the occurrence of compound heterozygosity for mutations in each of these genes as disease-causing events. No functionally relevant sequence variation in the coding sequence and flanking intronic regions of DHCR7 (MIM 602858) and KAT6B (MIM 605880), which are mutated respectively in Smith–Lemli–Opitz and Ohdo syndromes, was identified (27×, mean depth coverage). Overall, these findings strongly pointed to UBE3B as the gene mutated in KOS.

{kind=link}
{kind=link}
Truncating and missense mutations in UBE3B cause Kaufman oculocerebrofacial syndrome. (A) Chromatograms documenting homozygosity for the nonsense mutations identified in the two affected individuals included in this study are reported together with the respective reference sequences. (B) Location of disease causative mutations. The UBE3B gene includes 26 coding exons. The 5′- and 3′-untranslated regions are shown in black. Exonic regions coding for the IQ motif and the HECT domain are reported in blue and orange, respectively. The mutations identified in the present study are shown above the diagram, while those reported by Basel-Vanagaite et al12 are listed below. Homozygosity was documented in four unrelated subjects (black), while compound heterozygosity was reported in two siblings (red).
To confirm the causal involvement of UBE3B in the disorder, the entire coding sequence of the gene was scanned for mutations in DNA obtained from circulating leucocytes from a second, unrelated subject with clinical features fitting the trait (UCSC-KS02), whose apparently non-consanguineous family originated from a small (400 inhabitants), possibly endogamous village of southern Italy. Mutation analysis permitted the identification of a different homozygous nonsense lesion, c.1166G>A, predicting premature termination of the protein at codon 389 (p.Trp389stop) (figure 2A). Genotyping of parental DNAs demonstrated the heterozygous conditions for the truncating lesion, while the nucleotide substitution was not observed to occur in public databases as well as in the control DNA group (see above) scanned by high resolution melting analysis and direct sequencing. Similar to what was observed for the truncating c.556C>T change, the nonsense nucleotide substitution affected a region that was located upstream of the catalytic domain, thus leading to loss of UBE3B's ubiquitin ligase function in the patient.
To assess more precisely the phenotypic variability associated with mutations of UBE3B, scanning of the entire coding sequence of the gene was performed on a selected cohort of subjects with features overlapping, in part, those recurring in KOS (see online supplementary table S2). These subjects exhibited a unique combination of features recurring in KOS, and none of them showed a phenotype fitting Ohdo syndrome or any other disorder considered in the differential diagnosis of KOS. Mutation analysis was also performed on the patient originally described by Briscioli et al,6 for whom a diagnosis of KOS was proposed on the basis of co-occurrence of cognitive deficits, suggestive facial features (microcephaly, long and narrow face, sparse eyebrows, upslanting palpebral fissures, abnormal columella, and unusual nasal configuration), ocular anomalies, long and thin hands and feet, together with neonatal respiratory difficulties. The phenotype, however, was distinctive for the lack of blepharophimosis, reduced corneal diameter, ear anomalies, micrognathia, and significant neonatal feeding problems (see online supplementary table S2). No disease-causing mutation was identified in this group of patients, suggesting that UBE3B lesions were likely to be associated with a relatively homogeneous phenotype.
While this work was under review, biallelic mutations in UBE3B were reported by Basel-Vanagaite et al12 in four subjects from three unrelated families who were described as presenting a previously unrecognised, autosomal recessive entity characterised by blepharophimosis, facial dysmorphism, microcephaly, cognitive deficits, and reduced growth. Consistent with the present findings, three of the four reported UBE3B mutations were truncating (figure 2B), supporting the idea of UBE3B loss of function as the shared molecular event underlying their phenotype. Extensive clinical information was available for the six UBE3B mutation-positive patients (table 1). Overall, these subjects presented with a remarkably homogeneous phenotype that undoubtedly fits the condition originally described by Kaufman et al.1–4 From the analysis of the published and present records, the recognisable craniofacial appearance of KOS is characterised by microbrachycephaly, narrow and upward oriented palpebral fissures, epicanthus, telecanthus, a distinctive pattern of the eyebrows that originate under the orbital arch and are directed upward, becoming wide and sparse especially in the third lateral, small nose with anteverted nostrils, long philtrum, thin lips, and small mouth.
Clinical presentation of subjects with biallelic inactivating UBE3B gene mutations
Micrognathia is a constant feature, and usually associated with a high, narrow or cleft palate. The teeth are small, poorly formed, frail, and with multiple caries. Ear features include a cup shape with hypoplastic lobe and folded helix, stenosis of the external meatus, and preauricular tags. Microcornea, strabismus, myopia, and pale optic disk are frequently observed; nystagmus may also occur. A bell shaped thorax with telethelia is commonly observed, and fingers are generally long and thin with hyperextensibility at the metacarpophalangeal joints. The lower limbs are frequently involved with coxa valga and metatarsus varus in both feet. The natural history of KOS also appears rather constant, particularly during the neonatal period, with respiratory distress and laryngeal stridor, and growth retardation—the latter mainly ascribed to feeding difficulties due to poor sucking, pylorospasm and gastro-oesophageal reflux. Delayed psychomotor/language development and moderate to severe cognitive deficits are almost invariably observed; hypotonia is common. The gait is clumsy, crawling with widened basis but without signs of ataxia. Febrile seizures may occur without a significant electroencephalogram pattern. Affected subjects generally present a very similar behaviour with sociable and good natured disposition. Consistent with the observation of hypocholesterolaemia in three of the four UBE3B mutation-positive subjects reported by Basel-Vanagaite et al,12 low concentrations of total/high density lipoprotein cholesterol were documented in patient UCSC_KS02, confirming an underlying defect in cholesterol metabolism possibly resulting in decreased cholesterol synthesis.12
Discussion
We report here that UBE3B loss of function underlies KOS. UBE3B is a still functionally uncharacterised E3 ubiquitin-protein ligase. It is widely expressed during development as well as in adult tissues. Specifically, UBE3B has been reported to be highly expressed in the central nervous system (eg, cerebral cortex and cerebellum), digestive tract (eg, oesophagus and stomach), respiratory system (eg, nasopharynx and bronchi), as well as in multiple cell lineage of skin and other soft tissues (fibroblasts, chondrocytes, and epithelial cells) (http://www.proteinatlas.org/ENSG00000151148/; http://www.ebi.ac.uk/gxa/)12. While the functional characterisation of the E3 ligase properties of UBE3B in terms of substrate specificity and cellular pathways is required to appreciate the molecular mechanism(s) implicated in perturbation of the developmental processes promoted by impaired UBE3B function, mutation data indicate that the disease-causing lesions selectively interfere with the catalytic function of the protein. Among the six reported disease-associated mutations, five different nonsense, frameshift, and splice site mutations are predicted to cause protein truncation before or at the beginning of the HECT domain (figure 2B), and consequently to abolish UBE3B's E3 ligase activity. Consistent with this finding, Ube3b−/− mice exhibit a severely reduced bodyweight and size, decreased growth rate, muscular hypotonia, mild hearing impairment, and a reduced total cholesterol value, recapitulating several key features of KOS.12 Of note, human UBE3B encodes multiple gene products, which result from alternative transcript processing. Among these, two mRNA species contain alternative 3′ coding exons, resulting in products with a distinct, truncated C-terminus lacking the HETC domain. One of these truncated isoforms has been hypothesised to retain its ability to bind to substrates and possibly work as a negative regulator of UBE3B's ubiquitin ligase function.11 While alternative splicing in UBE3B has been proposed to possibly represent a novel fine-tuning mechanism in regulating activities of HECT domain ligases, the present observation that heterozygosity for a catalytically impaired UBE3B mutant does not result in any noticeable clinical phenotype and apparently does not operate with a dominant negative effect is contrary to this idea.
Analysis of the available clinical records supports the view that biallelic inactivation of UBE3B underlies the oculocerebrofacial syndrome originally recognised by Kaufman et al.1 While further investigations are required to define more precisely the molecular diversity of disease-causing mutations as well as the clinical spectrum associated with UBE3B mutations, screening of the entire coding sequence of the gene in a clinically heterogeneous cohort of patients sharing, in part, clinical features with KOS did not reveal any mutations. This finding suggests that loss of UBE3B catalytic function is associated with a clinically complex but relatively homogeneous and recognisable disorder in which anomalies involving the central nervous system (developmental delay, cognitive deficits, and hypotonia), cranium and face (microcephaly, distinctive eyebrow pattern, blepharophimosis, telecanthus, small nose with anteverted nostrils, long philtrum and small mouth), eyes (small cornea/microcornea, strabismus, myopia and pale optic disk), derivatives of the first and second pharyngeal arches (micrognathia, high and narrow/cleft palate, auricular stenosis, and preauricular tags), and other functional issues (neonatal respiratory difficulties, feeding and gastrointestinal problems and postural anomalies) co-occur as cardinal features. This conclusion is further supported by the data reported in the literature by Basel-Vanagaite et al,12 published while this work was under revision, which allows KOS to be defined clinically more precisely.
Ubiquitination is a widely used post-translational process employed to tag proteins for their degradation or intracellular sorting, as well as to modify their function directly.13 ,14 This protein modification has emerged as a critical mechanism involved in the regulation of a wide array of cellular processes, including cell division and differentiation, as well as development, with as many as 30–40% of all proteins estimated to be ubiquitinated in mammalian cells.15 Ubiquitination involves three classes of enzymes (E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes and E3 ubiquitin-protein ligases) that are required to catalyse ubiquitin activation and assembly of ubiquitin monomers or chains onto substrates. Among these, the E3 ubiquitin ligases constitute the largest group, including several hundred enzymes of various classes.16 By coupling substrate binding and ubiquitin transfer, E3 enzymes control the efficiency and specificity of protein ubiquitination. Within this group, the HETC domain-containing E3 ligases constitute a modest sized family of proteins defined by a conserved C-terminal catalytic domain and a variable N-terminal region conferring the ability to bind specifically to their substrates.17 Defective function of this class of E3 ligase has been documented to affect development. Loss of function of UBE3A (MIM 601623) has been identified to underlie Angelman syndrome (MIM 105830), a neurodevelopmental disorder characterised by severe cognitive deficiency, speech impairments, epilepsy, distinctive ataxic movements, abnormal sleep patterns, and hyperactivity.18 ,19
Other E3 ligases have been implicated in the pathogenesis of human genetic diseases, including von Hippel–Lindau disease (MIM 193300) (VHL, MIM 608537),20 autosomal recessive juvenile Parkinson disease (MIM 600116) (PARK2, MIM 602544),21 3M syndrome (MIM 273750) (CUL7, MIM 609577),22 Johanson–Blizzard syndrome (MIM 243800) (UBR1, MIM 605981),23 autoimmune polyendocrine syndrome type 1 (MIM 240300) (AIRE, MIM 607358),24 and the CBL (MIM 165360) mutation associated RASopathy (MIM 613563).25–27 The present findings provide further evidence of the dramatic impact of misregulation of protein ubiquitination on developmental processes. Notably, a key aspect of KOS is the involvement of structures that derive from the first and second pharyngeal arches. These embryonic domains, also known as branchial arches, are composed of mesenchymal cells of mesodermal and cranial neural crest origin, and give rise to a wide variety of skeletal, muscular, and neural elements of the face.28 Commitment, survival, migration, expansion, and differentiation of these cells is coordinated by a complex signalling network, whose deregulation has an impact on the development of many of the structures of the face.29 ,30 The abrogated function of UBE3B in KOS for the first time documents the relevant role of ubiquitination mediated processes in controlling developmental programmes of derivatives of these embryonic domains.
Acknowledgments
We are indebted to the families who participated in the study and Serenella Venanzi (Istituto Superiore di Sanità, Rome, Italy) for experimental support. We thank BGI (Hong Kong) for the high quality raw sequencing data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
MT and GZ contributed equally.
-
Contributors EF and VF performed biological sample processing, sequence data validation, segregation analyses, and mutation screening; AC and VC performed the bioinformatics analyses of exome sequencing data; CL, DM, MFB, LM, AP and GZ recruited patients, collected biological samples, and performed clinical evaluations; EF, MT and GZ wrote the manuscript; MT and GZ conceived the study and analysed the data.
-
Funding Istituto Superiore di Sanità (ricerca corrente 2012) and Telethon-Italy (GGP10020) to M.T.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Istituto Superiore di Sanita’, Rome, Italy.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Web resources Accession numbers and the URL for data are as follows:
-
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
-
1000 Genomes Project variant database, http://www.1000genomes.org/
-
wAnnovar, http://wannovar.usc.edu
-
Picard's MarkDuplicates utility, http://picard.sourceforge.net
-
Entrez Gene, http://www.ncbi.nlm.nih.gov/gene/
-
Simple Modular Architecture Research Tool (SMART), http://smart.embl-heidelberg.de/
-
Gene Expression Atlas, http://www.ebi.ac.uk/gxa/
-
Human Protein Atlas, http://www.proteinatlas.org/ENSG00000151148/.