Article Text

Abstract
Over 15 years have passed since the discovery of the first autoinflammatory gene, MEFV, responsible for familial Mediterranean fever. The identification of another gene, TNFRSF1A, in 1999 led to the concept of autoinflammation which characterises rheumatological conditions triggered by a defective innate immunity. Substantive progress has been made since then with the identification of 18 autoinflammatory genes accounting for up to 24 disease entities showing overlapping symptoms. The accumulation of studies reporting patients with missing or excess mutations as compared with expected numbers favours the hypothesis that these diseases are distributed along a continuum ranging from monogenic to multifactorial conditions, rather than featuring only classical modes of inheritance. Moreover, the probable interactions of environmental and epigenetic factors further obscure our understanding of the mechanisms underlying the phenotypic expression of patients. This review explores the history of autoinflammatory gene discovery, discusses the nosological disparities stemming from the clinical versus pathophysiological definition of autoinflammatory diseases and summarises various inheritance patterns. This review calls for a consistent disease nomenclature and presents a reconciling hypothesis which places different sequence variants within the autoinflammatory disease continuum. Integrating these new concepts should help to facilitate communication between health professionals and promote personalised patient care.
- Genetics
Statistics from Altmetric.com
Introduction
The name autoinflammation was proposed in 1999 to echo that of autoimmunity.1 ,2 However, the rather artificial boundaries between autoinflammatory diseases (innate immunity) and autoimmune diseases (adaptive immunity) are gradually disappearing to give rise to the currently preferred concept of immunological disease continuum.3 Behçet's disease is a good example of a multifactorial condition with features of both autoinflammation and autoimmunity.4 ‘Pure’ autoinflammatory diseases are classically defined as conditions without high titre auto-antibodies or antigen-specific T cells. While initially focused on hereditary recurrent fevers (HRFs), autoinflammatory diseases now encompass a wide spectrum of disorders ranging from rare monogenic, for example, the prototype familial Mediterranean fever (FMF), to more frequent multifactorial diseases, for example, Crohn's disease. This explains why the team which originally proposed the term ‘autoinflammatory disease’ now suggests that its definition should be reassessed to include all clinical disorders marked by abnormally increased inflammation, mediated predominantly by the innate immune system, with a significant host predisposition.5 Indeed, most cases of autoinflammatory disease are characterised by attacks of systemic inflammation resulting in recurrent fever, acute arthritis and increased acute phase reactants. Frequent accompanying symptoms are skin lesions or rashes, myalgia, bone deformation or sensorineural defects. The key symptoms of autoinflammatory diseases are listed in table 1. The appropriate diagnosis of a disease can prove challenging due to overlapping or mimicking symptoms, or even association with, autoimmune diseases. Although several classifications of autoinflammatory diseases have been proposed in order to help the clinician, not one of these has been consensually adopted (box 1).
Evolving classification of autoinflammatory diseases
-
The first classifications of autoinflammatory diseases were based on the patients’ phenotype. Hence, hereditary recurrent fevers including the prototype familial Mediterranean fever define monogenic disorders with recurrent fever as the key symptom.
-
More recently, McGonagle and colleagues3 suggested an integrated classification using the autoimmune versus autoinflammatory ratio involved in the condition as a means to rank each autoinflammatory condition within the continuum of immunological diseases.
-
At the same time, the NIH group suggested a classification scheme based on the molecular insights gathered over the past decade.88
-
No authoritative classification has emerged in 2012, most likely due to the extending identification of phenotype overlaps and genetic heterogeneity.
Genes responsible for monogenic autoinflammatory diseases and their corresponding mode of transmission, disease names and key symptoms
Our understanding of the pathophysiology of autoinflammatory diseases has dramatically increased due to the unravelling of defective signalling pathways. The discovery of the FMF gene in 1997 shed light on a new death domain (the pyrin domain6) that presents high homology with proteins involved in inflammation and apoptosis.7 ,8 A second gene containing a death domain was found in 1999 to be responsible for a dominant form of recurrent fever: Tumour Necrosis Factor (TNF) Receptor Associated Periodic Syndrome or TRAPS.2 Since then, 16 other autoinflammatory genes have been discovered (table 1), including more than 1000 sequence variants.9–11 Their clinical significance is often unclear, and the mode of transmission of these diseases is becoming difficult to interpret as the number of families with unexpected genetic associations or patterns of inheritance is growing.
This review has compiled comprehensive genetic data on autoinflammatory diseases in order to clarify the current complexity and controversy surrounding their inheritance, and calls for a consistent disease nomenclature. A list of current disease and gene acronyms is provided in box 2.
Acronyms in autoinflammatory diseases
-
APLAID, Autoinflammatory PLCG2-associated Antibody deficiency and Immune Dysregulation
-
CANDLE, Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature syndrome
-
CAPS, Cryopyrin Associated Periodic Syndrome
-
CARD14, Caspase Recruitment Domain Protein 14
-
DIRA, Deficiency of Interleukin 1 Receptor antagonist
-
DITRA, Deficiency of Interleukin 36 Receptor antagonist
-
DSAP, Disseminated Superficial Actinic Porokeratosis
-
FCAS, Familial Cold Urticaria
-
FMF, Familial Mediterranean Fever
-
HIDS, Hyper IgD Syndrome
-
HOIL-1, Haeme-Oxidised IRP2 ubiquitin Ligase 1
-
HRF, Hereditary Recurrent Fever
-
IBD, Inflammatory Bowel Disease
-
IL10, Interleukin 10
-
IL10RA, Interleukin 10 Receptor α
-
IL10RB, Interleukin 10 Receptor β
-
IL1RN, Interleukin 1 Receptor Antagonist
-
IL36RN, Interleukin 36 Receptor Antagonist
-
JMP, Joint Contractures, Muscular Atrophy, Microcytic Anaemia and Panniculitis-Induced Lipodystrophy
-
LPIN2, Lipin2
-
MEVA, Mevalonic Aciduria
-
MEFV, MEditerranean FeVer
-
MKD, Mevalonate Kinase Deficiency
-
MVK, MeValonate Kinase
-
NAPS12, NLRP12-Associated Periodic Syndrome
-
NLRP12, NOD-Like-Receptor Family, Pyrin Domain-Containing 12
-
NLRP3, NOD-Like-Receptor Family, Pyrin Domain-Containing 3
-
NNS, Nakajo-Nishimura Syndrome
-
NOD2, Nucleotide-binding Oligomerisation Domain Protein 2
-
PAPA, Pyogenic sterile Arthritis, Pyoderma gangrenosum, and Acne
-
PLAID, PLCG2-associated Antibody deficiency and Immune Dysregulation
-
PLCG2, Phospholipase C, γ-2
-
PRP, Pityriasis Rubra Pilaris
-
PSMB8, Proteasome Subunit, β-Type, 8
-
PSORS, Psoriasis
-
PSTPIP1, Proline/Serine/Threonine Phosphatase-Interacting Protein 1
-
RBCK1, Ranbp-Type and C3HC4-Type Zinc Finger-Containing 1
-
SH3BP2, SH3 Domain-Binding Protein 2
-
TNFRSF1A, Tumour Necrosis Factor Receptor Super Family 1A
-
TRAPS, TNF Receptor Associated Periodic Syndrome
-
VUS, Variant of uncertain significance
Lessons from HRFs
HRFs are the prototype of autoinflammatory diseases as they include the first disease whose gene was identified, that is, FMF.7 ,8 While HRFs are classically monogenic families with unexpected genetic patterns have been reported (box 3) and are discussed below. Large series of patients with recurrent fevers and biological inflammation, in which other common inflammatory diseases have been ruled out, are now available enabling the dissection of genetic and non-genetic factors affecting the patient phenotype.
Genetic mechanisms involved in the inheritance of hereditary recurrent fevers
-
Complex allele: the combination of two or more mutations occurring in cis within the same gene
-
Gene dosage: effect on the phenotype is proportional to the number of mutated alleles
-
Genetic heterogeneity: mutation(s) in one of alternative genes with a strong effect causing the disease
-
Genetic homogeneity: mutation(s) in a single known gene with a strong effect causing the disease
-
Modifier genes: genes responsible for variable expression of the disease
-
Oligogenism: mutation(s) in a limited number of genes with an intermediate effect causing the disease (two genes acting together cause digenism)
-
Pseudodominance: vertical segregation of recessive diseases due to abnormally high mutation frequency usually related to consanguinity or founder effect
-
Susceptibility genetic factors: several genes with a mild effect act together to cause the disease
Missing mutations
This chapter includes the discovery of either no mutations in the suspected gene or of a single mutation in recessive diseases. Such patients are hereafter referred to as ‘genetically orphan’. Several scientific and medical reasons could account for the 20%–85% of genetically orphan patients, including the large number of autoinflammatory genes presumed to be unidentified, the lack of functional tests (except for mevalonate kinase deficiency (MKD)) and the rather unspecific main clinical feature (recurrent fever at the paediatric age) which characterises most autoinflammatory patients.12–19 However, this significant proportion could also be due to the evolution in how many of the autoinflammatory diseases are defined, which remains a matter of debate and depends on the habits and discipline of health professionals.
Familial Mediterranean fever
FMF was known as periodic disease or recurrent polyserositis, prior to being called ‘Familial Mediterranean fever’ by Heller in 1955,20 a name now universally used. All of these names clearly refer to the key symptoms of the disease. However, fever and polyserositis are also common to the other HRFs. Livneh and others defined the clinical criteria for FMF.21–24 Although very useful to the clinicians, they could not be totally specific. As numerous patients with FMF clinical criteria but without MEFV mutations were reported, genetic heterogeneity was proposed.25 However, since then a MEFV2 gene has not been identified (figure 1), and the proportion of genetically orphan FMF patients depends on patient ancestry. It is low in Mediterranean countries (20%–25%), although very high in other populations (80%–85%).16 ,26 This high rate may also be due to wrong clinical overdiagnosis. FMF is classically recessively transmitted, yet cases of dominant fever were reported which justified the creation of a new entity named ‘dominant periodic fever’ as no genetic test was available until 1997. After this milestone, some families were indeed related to the MEFV gene since a dominant segregation of a heterozygous MEFV mutation was proven, and series of FMF patients with a single MEFV mutation were described.27–33 After an unsuccessful search for the second mutation, it was speculated that a true dominant transmission of certain severe mutations could exist. A recent study derived from the largest European cohort, Eurofever, a shared online registry for patients with autoinflammatory diseases,34 ,35 supported gene dosage.36 Gershoni and colleagues suggested that complex alleles can lead to a more severe form of the disease.37 ,38 Among other hypotheses are as yet unknown molecular mechanisms, such as regulation of transcription,39 non-exonic mutations40 ,41 or, more likely, mutations in another gene.25 ,42–45
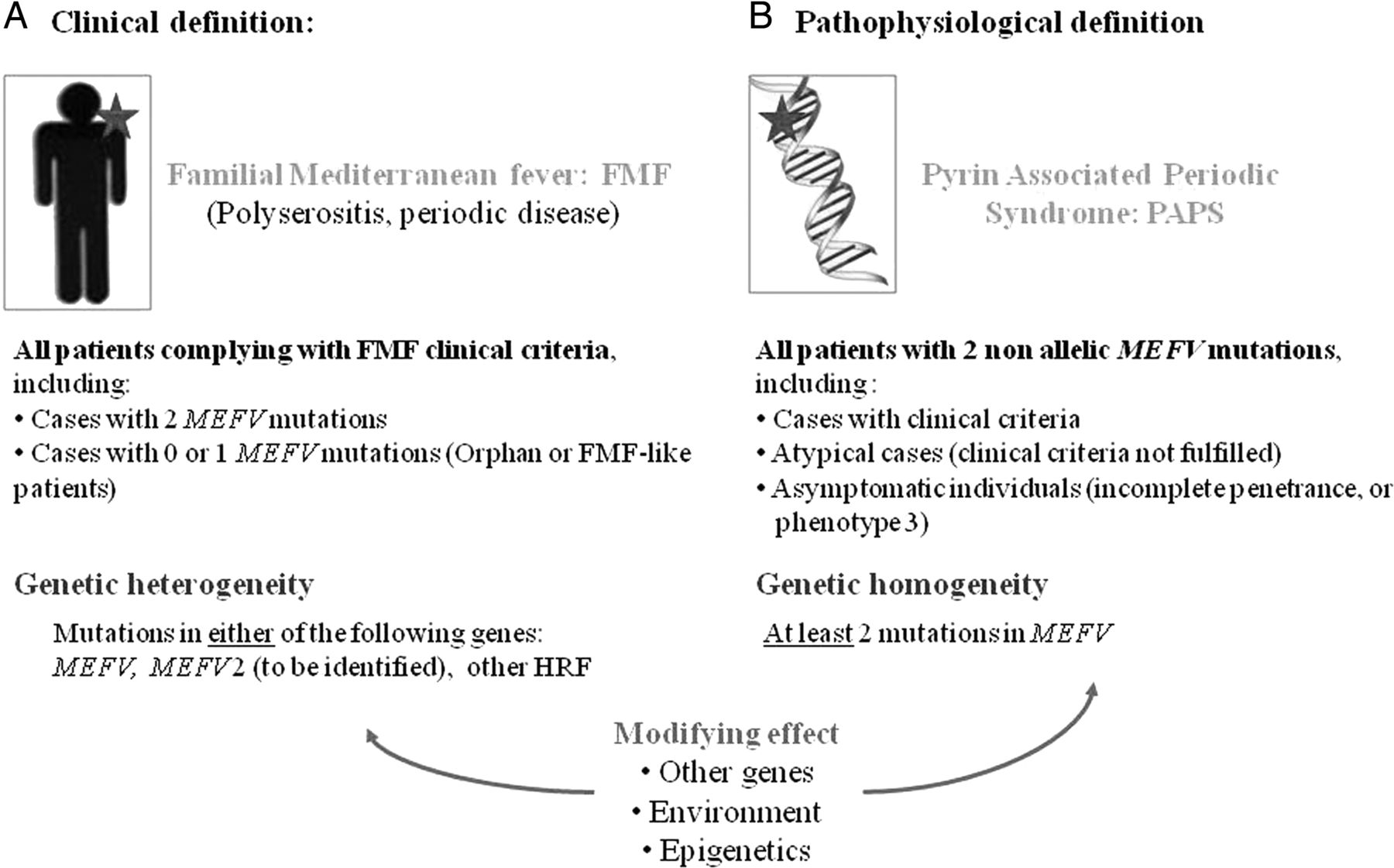
Clinical versus pathophysiological definition of autoinflammatory diseases; discussion on the example of familial Mediterranean fever (FMF). (A) Clinical definition: FMF currently has a clinical name. As it is a recessive disease, two mutations in the causative gene MEFV are expected. Provided that the gene has been totally explored and rare mutations excluded, which is never routinely carried out, there are two hypotheses accounting for those patients with nil or only one mutation: existence of another gene (MEFV2) or reassignment of the patients to another hereditary recurrent fever (HRF). Other genetic, environmental or epigenetic factors may modify the phenotype although are not mandatory for the disease to occur. Multifactorial diseases mimicking FMF are not discussed. (B) Pathophysiological definition: A naming proposition matching other HRFs. Pyrin Associated Periodic Syndrome would cover all patients with two non-allelic MEFV mutations. Phenotype 3 defines asymptomatic individuals.114 This figure is only reproduced in colour in the online version.
TNF Receptor Associated Periodic Syndrome
In 1968, there were reports of a Swedish family with recurrent fever, abdominal pain that persisted for 7–14 days and renal deposition of amyloid identical to that seen in FMF.46 In 1982, another family with an autosomal dominant periodic fever was described in Ireland, and the term ‘Hibernian fever’ was created on this occasion.47 It was revealed that these families and others identified later48 ,49 suffered from genetic conditions other than FMF. Upon the 1999 discovery of the causative gene, TNFRSF1A, McDermott et al2 preferred to associate the phenotype with the mutated protein, and created the TRAPS acronym that stands for ‘TNF Receptor Associated Periodic Syndrome’. ‘Hibernian’ or ‘dominant periodic’ fevers are no longer in use. As a TNFRSF1A mutation was not identified in a proportion of patients with the TRAPS-like phenotype, Borghini et al44 analysed the sequence of genes known to play a pivotal role in the mechanisms downregulating the TNF-induced inflammatory response such as receptor shedding, receptor internalisation and negative regulation of nuclear factor-κB transcription. This work revealed only a few unreported variants, apparently neutral; therefore, the authors concluded that the gene(s) responsible for TRAPS-like syndrome still remain to be identified.
Cryopyrin Associated Periodic Syndrome
A similar logic was applied to name the three clinical entities caused by dominant mutations within the same gene, NLRP3, encoding cryopyrin. ‘Cryopyrin Associated Periodic Syndrome’ (CAPS50 or cryopyrinopathies51) was generated as a unifying term for Familial Cold Autoinflammatory Syndrome (FCAS), Muckle-Wells Syndrome and Chronic Infantile Neurological Cutaneous and Articular syndrome, also known as Neonatal-Onset Multisystem Inflammatory Disease. These conditions are no longer considered as three subphenotypes with increasing severity because of the heterogeneous presentation of this monogenic disease. Instead, the indepth examination of patients with NLRP3 mutations demonstrated that they display a continuum of symptoms.51 Consistently, although some mutations were associated with only one of the three CAPS diseases, for example, L305P in FCAS, several sequence variants did not clearly correlate with the phenotype.51 The absence of clear dominant segregation was explained in some families by the existence of de novo mutations45 (also found in TRAPS52) and mosaicism.53–55 It was also revealed that some of the orphan CAPS patients had mutations in other genes, such as NLRP1256 or IL1RN.57
Mevalonate kinase deficiency
‘Hyperimmunoglobulinemia D and periodic fever Syndrome’ (HIDS, formerly Periodic Fever, Dutch Type58) owes its name to increased IgD titres reported in patients suffering from recurrent fever, macula-papular rashes and arthralgia lasting for 3–7 days. Active gene hunting unexpectedly revealed that this recessive condition was caused by mutations in the mevalonate kinase gene59 responsible for severe mevalonic aciduria. The switch to the pathophysiological name, MKD, is progressing slowly.58 All autoinflammatory experts agree that the term ‘HIDS’ should be dropped since high IgD sera levels are not specific and can be found in other inflammatory conditions60–62 or can be absent in MKD.59 ,61 ,63 ,64 MKD is classically recessively inherited and true orphan cases either do not exist or are scarce, since the rate of genetic confirmation (homozygosity or compound heterozygosity) nearly reaches 100% in patients with decreased mevalonate enzymatic activity.65 Unexpectedly, dominant mutations in this same gene were recently associated with ‘disseminate superficial actinic porokeratosis’ (DSAP).66 Although DSAP patients had no clinical features of MKD, or abnormalities in their serum IgD concentrations, it remains unclear whether they had decreased enzymatic activity. However, the mutations c.417_418insC and c.604G>A found in two DSAP patients were also reported in two different MKD patients in a compound heterozygous state.
Excess mutations
More than two mutations in recessive diseases
Consanguinity, as well as founder effect, is a well-recognised cause of pseudodominance in recessive diseases, as they increase the frequency of rare pathogenic alleles. It is not unusual that three mutations segregate in a single family suffering from FMF.67 One such case was also identified in a MKD family.68 Four mutations were found in Druze FMF patients.69
Two mutations in dominant diseases
A handful of reports describe patients bearing two mutations in either the gene TNFRSF1A or NLRP3, responsible for dominant HRFs.70–73 In most cases, one of the two sequence variants is of unclear significance (VUS), such as p.Arg121Gln (usual name R92Q, c.362G>A,) or p.Val198Met (V198M, c.592G>A), in these two genes respectively. Three R92Q homozygotes were identified (ref70 and personal communication), and one p.Pro75Leu (usual name P46L, c.224C>T) homozygote (personal communication). Other combinations include V198M and p.Leu353Pro (L353P, c.1058T>C) within NLRP3 in a patient with a FCAS phenotype,73 and p.Cys102Trp (usual name C73W, c.306C>G) and R92Q within TNFRSF1A in a patient with a TRAPS phenotype (personal communication). It is difficult to determine whether there is a gene dose response as first, the variants are usually not phased and may well be located on the same chromosome (complex allele), and second, the reported cases are too scarce to allow statistical evaluation.
Mutations in more than one gene
Cases in which several mutated genes cluster are increasingly reported (figure 2), confusing the clinician while sometimes regrettably delaying accurate and relevant patient care. Up to four mutations have been found. The most likely cause of association is a fortuitous coexistence of highly prevalent mutations in a single individual. Indeed, in most patients one of the two mutations was common in the general population. Digenism was also investigated;29 ,72 however, this was only confirmed in one case through clearly demonstrating cosegregation of the R92Q and V198M variants with the phenotype in a multiplex family suffering from a dominant form of autoinflammatory disease with features of TRAPS, CAPS and Behçet's disease.72

Dual mutations in hereditary recurrent fever (HRF) genes. A colour code is used to represent the four main HRF genes: Blue: MEFV; Green: TNFRSF1A; Red: MVK; Orange: NLRP3. Darker backgrounds indicate patients with confirmed genetic diagnosis: two clearly pathogenic mutations for recessive diseases (familial Mediterranean fever (FMF) and mevalonate kinase deficiency (MKD)), and one clearly pathogenic mutation for dominant diseases (TNF Receptor Associated Periodic Syndrome and Cryopyrin Associated Periodic Syndrome). The number of patients recorded with a given genotype is proportional to the surface of the corresponding circle. Overlaps depict the coexisting variants reported in the two genes. The most frequent combinations tend to include variants of unclear significance such as E148Q (MEFV) and R92Q (TNFRSF1A) as defined in the guidelines for the genetic diagnosis of HRFs.115 M694V and p.Val377Ile (V377I, c.1129G>A) are the two most frequent causative mutations in FMF and MKD, respectively. This figure is only reproduced in colour in the online version.
Modifying factors
Genetic factors
Gender and certain mutations are known to confer a higher risk of renal amyloidosis to patients. p.Met694Val (M694V, c.2080A>G) homozygosity has been repeatedly associated with severe disease and kidney complications in FMF patients.26 In addition, cysteine mutations in the TRAPS gene are thought to lead to renal amyloidosis, although large series are lacking.74 Polymorphisms in the major histocompatibility complex class I chain-related gene A (MICA)75 ,76 and the serum amyloid-A genes75 ,77–79 have both been reported as bad prognostic variables in FMF.
Environmental factors
Environment has been suspected as a modifying factor since the initial study of Armenian FMF patients living in California.80 The authors found that migrants had less amyloidosis than the other patients. In a very large collaborative study,81 we assessed several factors that may play a role in renal amyloidosis, and showed that the country of recruitment, rather than MEFV genotype, was the key risk factor. This risk paralleled infant mortality rates, a highly sensitive indicator of population health which reflects economic development, general living conditions, social well-being and quality of environment.81 Ozen et al82 showed that 78% of patients living in Turkey had a severe course of FMF compared with 34% living in Germany. Based on the concordance in clinical manifestations in Israeli monozygote and dizygote twins, Ben-Zvi et al83 calculated that the environmental effect on the phenotype of FMF was estimated at around 12%.
Epigenetic factors
Very little is known about the involvement of epigenetics in autoinflammatory disorders. In the sole available study, the methylation level of MEFV exon 2 within tended to be slightly higher in FMF patients compared with healthy controls.84 This needs further confirmation in larger patient series and in other populations.
Future directions
How to deal with new autoinflammatory diseases?
Issue: phenotype overlaps, name complexity
The tremendous insights gained into the pathophysiology of autoinflammatory diseases fuelled candidate gene screening and exome sequencing in genetically orphan patients. This research confirmed that a single gene could be involved in various phenotypes and, conversely, many different genes could be involved in overlapping phenotypes (table 1).
Three disease names were developed for a group of phenotypes caused by mutations in the Proteasome Subunit, β-Type 8 (PMSB8) gene. These names reflect the main clinical features, that is, ‘joint contractures, muscle atrophy, microcytic anaemia and panniculitis-induced lipodystrophy syndrome’,85 or ‘chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome’,86 or the name of the physicians who described the disease at that time, ‘Nakajo-Nishimura syndrome’.87
Families with cold-induced urticaria and fever yet no NLRP3 mutations (orphan CAPS) were investigated. Two of them were related to mutations in NLRP12. This condition has no consensual name. It was still unnamed upon the gene discovery,56 and was agreed as NLRP12 Associated Periodic Syndrome to parallel TRAPS and CAPS in a database dedicated to autoinflammatory mutations, Infevers (http://fmf.igh.cnrs.fr/ISSAID/infevers/).9 Later, it was labelled FCAS2 or Guadeloupe variant periodic fever in a review by Masters and coworkers,88 as well as NLRP12 associated disorders by the gene discoverer in 2011,89 and alternatively FCAS2, NLRP12 associated disease and NLRP12-related periodic fever in two recent papers by the Eurofever consortium.35 ,90
The pathophysiological rule was adopted to name two recently recognised recessive autoinflammatory conditions with severe skin lesions, deficiency of interleukin 1 receptor antagonist (DIRA)57 and deficiency of interleukin 36 receptor antagonist (DITRA).91 However, a naming issue could arise if mutations in another member of the interleukin receptor antagonist family also happened to be responsible for an autoinflammatory disease. The DIRA gene was identified after a candidate approach in three families negative for NLRP3 mutations, but fully responsive to anti-interleukin 1 therapies, while DITRA was discovered after exome sequencing of consanguineous Tunisian families with generalised pustular psoriasis. A recent paper showed that the DITRA spectrum also covers palmar-plantar pustulosis and acrodermatitis continua of Hallopeau.92
The psoriasis susceptibility locus 2 (PSORS2) gene was revealed to be responsible for a second monogenic form of psoriasis caused by mutations in the dominant gene, caspase recruitment domain family, member 14 (CARD14).93 ,94 CARD14 is also responsible for familial pityriasis rubra pilaris.95
Autoinflammatory phospholipase C, γ 2 (PLCγ2) associated antibody deficiency and immune dysregulation (APLAID) is caused by dominant mutations in the PLCγ2 gene.96 This syndrome is the autoinflammatory form of PLCγ2 associated antibody deficiency and immune dysregulation (PLAID, also called FCAS3).97
The latest hereditary autoinflammatory syndrome which has been linked to a gene has not been named.98 ,99 The patients with this unnamed syndrome presented with chronic inflammation, bacterial infections and muscular amylopecinosis. They were found to carry biallelic loss-of-expression and loss-of-function mutations in RBCK1 (HOIL1), a component of the linear ubiquitination chain assembly complex.
Solution: disease nomenclature consistency
Ben-Chetrit and colleagues suggested the adoption of the general name ‘pyrin associated disorders’ for FMF.100 An approach consistent with TRAPS and CAPS would call for ‘PAPS’, standing for Pyrin Associated Periodic Syndrome (figure 1). There are pros and cons in generalising the nomenclature in this way.
The advantages are that all patients diagnosed with protein associated disease names would de facto be genetically confirmed. The notion of genetic heterogeneity would no longer have justification. Any existing clinical criteria would be supportive, but dispensable when unavailable. The best example is TRAPS, the HRF with the largest clinical spectrum for which there have been no attempts to delineate clinical criteria. Interestingly, this rule unifies the genetic aspect and catalyses the development of targeted and therefore more efficient therapeutic molecules.
Drawbacks of using this paradigm include the mathematically higher proportion of genetically orphan patients with a convincing autoinflammatory phenotype, coined with the inelegant yet tempting diagnosis of ‘autoinflammatory disease-like patients’. More of an issue is the fact that the definition of ‘mutation’ is becoming vague for sequence variants that were thought to be causative upon the gene discovery, yet are now believed to be at best functional polymorphisms. Classical examples are p.Glu148Gln (E148Q, c.442G>C) in the MEFV gene and R92Q in the TNFRSF1A gene. Guidelines have been assembled to help genetic interpretation of these variants. The group of patients with a ‘low penetrance’ genotype would also increase, especially if mutational screening of asymptomatic or atypical relatives is boosted with the development of high throughput genetic techniques.
A consensus conference proved both successful and helpful for the nomenclature of proteins involved in innate immunity regulation.101 For example, the key inflammasome protein was given the alternative names of cryopyrin, CIAS1, NALP3, PYPAF1 and CTRL1.1 before it was definitively fixed as NLRP3. Such a meeting would be highly recommended to harmonise the names of current and future autoinflammatory monogenic conditions.
How to deal with VUS?
Issue: monogenic versus multifactorial diseases
An increasing number of studies support the involvement of sequence variants of genes responsible for Mendelian autoinflammatory diseases in either autoinflammatory or autoimmune complex conditions (figure 3). Crohn's disease is one of the first multifactorial diseases in which an innate immunity gene named NOD2/CARD15,102 responsible for a dominant autoinflammatory disorder named Blau,103 was identified as a major inflammatory bowel disease (IBD1) gene. The FMF, TRAPS and CAPS genes were also inconsistently associated with Crohn's disease.104–107 MEFV and TNFRSF1A may have a role in Behçet's disease.43 ,108 MEFV, NLRP3 and TNFRSF1A were suggested as susceptibility factors for multiple sclerosis. The variant R92Q in the latter gene was the most convincing as it was confirmed in multiple genome-wide association studies.109 Nevertheless, this mutation did not appear to be a severity marker of the disease neither modifying the clinical progression of multiple sclerosis nor its therapeutic response. Although the NLRP3 inflammasome has a critical role in monosodium urate crystal-induced inflammatory gout attacks,110 no study demonstrated NLRP3 variants associated with gout. All in all, this review revealed numerous conflicting results (figure 3), probably due to different study designs and populations.

Association of genes responsible for monogenic autoinflammatory diseases with multifactorial diseases. To obtain the most relevant figures and avoid duplicated data, studies were only retained which described significantly large series. Reviews and case reports were not included. Studies consistently demonstrating significant association of the gene with the disease are depicted in red; studies showing a trend or contradictory results are shown in purple; blue is used for absence of association. AS, ankylosing spondylitis; BD, Behçet's disease; CAD, coronary artery disease; CD, Crohn's disease; CeliacD, celiac disease; FMS, fibromyalgia syndrome; HSP, Henoch-Schönlein purpura and polyarteritis nodosa; IBD, inflammatory bowel disease; JIA, juvenile idiopathic arthritis; MI, myocardial infarction; MS, multiple sclerosis; PBC, primary biliary cirrhosis; PFAPA, periodic fever with adenitis pharingitis and aphtosis; PR, palindromic arthritis; PsA, psoriasis; PsoJIA, psoriatic JIA; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; UC, ulcerative colitis. This figure is only reproduced in colour in the online version.
Solution: integrating patient background
The coexistence of complex inflammatory diseases and monogenic autoinflammatory gene variants common in the general population is likely to be fortuitous. An appealing hypothesis is that ‘severe mutations’ (eg, M694V in MEFV) could segregate with specific monogenic disorders, while other ‘mild/low penetrance mutations’ (eg, R92Q within TNFRSF1A) could favour non-specific inflammation (figure 4). It is tempting to speculate that the accumulation of such VUS associated with a non-genetic background could either trigger multifactorial diseases or at least modify their expression or severity. Depending on circumstances, a sequence variant could behave as a true causative mutation, a functional polymorphism or remain silent. This hypothesis reconciles the current controversies surrounding the clinical significance of several sequence variants such as E148Q (MEFV) or Q703K (NLRP3), and accounts for why they may be found at the same frequency in both patients and the general population.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inheritance of autoinflammatory diseases, a hypothesis bridging monogenic and multifactorial conditions. Autoinflammatory diseases are classically classified as either monogenic or multifactorial; however, many families do not fit into these models. This hypothesis proposes that the modes of inheritance of autoinflammatory diseases are distributed along a continuum involving mutation type, number of genes and environmental factors. VUS, variants of unknown significance. The thickness of the line depicts the strength of the environmental effect. This figure is only reproduced in colour in the online version.
Current genetic diagnostic strategies which are restricted to the screening of a limited number of genes or exons will not definitively resolve the issue of dealing with VUS. The hypothesis proposed will undoubtedly be confirmed with the development of exome sequencing approaches which will provide phenotype associated genetic signatures.
Conclusions
Increasing knowledge of the inheritance of autoinflammatory diseases switched simple paradigms into puzzling genetic situations. FMF, first described as a recessive disease due to a handful of clear causative mutations, was revealed to be possibly dominantly inherited and its gene to harbour a constellation of VUS (more than 20 publications for E148Q) associated with other inflammatory disorders such as juvenile idiopathic arthritis.111–113 This review demonstrated that at least part of the controversy relies on nomenclature. Prior to gene discovery, a clinical definition based on established criteria was generally assigned for most of the autoinflammatory diseases (FMF, FCAS, etc). However, the history of the names of the diseases reflects the trend towards a nomenclature which includes the name of the mutated protein. In some cases, a pathophysiological definition has been (re)assigned subsequent to gene discovery (eg, TRAPS, MKD). In parallel, large scale collaborative and country-specific studies confirmed recurrent unproved hypotheses, for example, the role of environment. Other questions such as the true existence of dominant FMF, the influence of epigenetic factors and oligogenism remain unanswered.
While the discovery of these genes opened up new ways of diagnosing these diseases through the molecular screening of mutations, the large accumulation of sequence data demonstrates that interpretation of genetic diagnosis and counselling is becoming increasingly difficult. This advocates the continuous efforts toward a network of multidisciplinary specialists, including molecular biologists, epidemiologists, clinicians, statisticians and web scientists, in order to: (i) elaborate registries devoted to gathering a critical mass of genetic and clinical data such as Infevers9–11 or Eurofever34 ,35 and (ii) promote high throughput sequencing which will improve patient care through better understanding of disease-specific defective signalling pathways and subsequent adaptation of patient therapies.
Acknowledgments
The author wishes to thank Joanne Strawbridge (Angloscribe) and V Macioce for English language editing and Drs L Cuisset, S Grandemange and M Barat for constructive reviewing of the paper.
References
Footnotes
-
Contributors IT is responsible for the overall content as both author and guarantor. IT had the idea for the article, performed the literature search and wrote the article.
-
Funding This work was supported by the French Ministry of Health and the Montpellier University Hospital Center.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.