Article Text

Abstract
Background Congenital multiple intestinal atresia (MIA) is a severe, fatal neonatal disorder, involving the occurrence of obstructions in the small and large intestines ultimately leading to organ failure. Surgical interventions are palliative but do not provide long-term survival. Severe immunodeficiency may be associated with the phenotype. A genetic basis for MIA is likely. We had previously ascertained a cohort of patients of French-Canadian origin, most of whom were deceased as infants or in utero. The goal of the study was to identify the molecular basis for the disease in the patients of this cohort.
Methods We performed whole exome sequencing on samples from five patients of four families. Validation of mutations and familial segregation was performed using standard Sanger sequencing in these and three additional families with deceased cases. Exon skipping was assessed by reverse transcription-PCR and Sanger sequencing.
Results Five patients from four different families were each homozygous for a four base intronic deletion in the gene TTC7A, immediately adjacent to a consensus GT splice donor site. The deletion was demonstrated to have deleterious effects on splicing causing the skipping of the attendant upstream coding exon, thereby leading to a predicted severe protein truncation. Parents were heterozygous carriers of the deletion in these families and in two additional families segregating affected cases. In a seventh family, an affected case was compound heterozygous for the same 4bp deletion and a second missense mutation p.L823P, also predicted as pathogenic. No other sequenced genes possessed deleterious variants explanatory for all patients in the cohort. Neither mutation was seen in a large set of control chromosomes.
Conclusions Based on our genetic results, TTC7A is the likely causal gene for MIA.
- Gastroenterology
- Genetics
- Developmental
- Molecular genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Hereditary multiple intestinal atresia (MIA) (MIM (243150)) is a severe congenital disorder, first formally defined in 1973.1 MIA typically involves multiple lesions which occur at various levels throughout the small and large intestines.2 Although surgical intervention is sometimes attempted, outcomes are poor and the condition is usual fatal within the first month of life. To date, no primary aetiology has been proved for the condition. Importantly, in some cases MIA is also associated with either mild or severe combined immunodeficiency (SCID),2–5 raising the possibility that an abnormal immune response might be the origin of the disease. However failure of normal embryogenesis remains a possibility with inflammation or immune dysfunction only secondary. Most MIA cases are sporadic, although a genetic basis for congenital cases is likely given some familial recurrences.2 ,6 A cluster of cases from Quebec, ascertained by us, is also consistent with a genetic cause, given frequent documented French-Canadian founder effects going back to the first immigrants and leading to increased incidences of specific recessive disorders.7 Other neonatal conditions involving local atresias within the small or large intestine are also known, but it remains unclear whether these represent fundamentally different disease entities from MIA or simply less severe examples.
Through whole exome sequencing of patients of French-Canadian origin, we identified two mutations in a single gene, the tetratricopeptide repeat (TPR) domain 7A gene TTC7A, that could explain the disease in the affected cases. Mouse mutations in the orthologous gene are known and while the mice exhibit some abnormalities in their immune system, they lack a gastrointestinal phenotype.8–10
Methods
Clinical ascertainment and patient descriptions
In the course of clinical practice over many years, more than 20 patients with MIA have been identified at the Centre Hospitalier de l'Université Laval (CHUL) in Québec City, some previously described.2 All cases were reportedly of French-Canadian origin, though not formally related by known genealogy up to their great-grandparents. DNA suitable for genome-wide analysis was obtained from these cases as well as three newly ascertained cases. DNA from three additional deceased cases was obtained from paraffin blocks (F2, F4 and F5). We also had intestinal tissues embedded in plastic for two deceased cases (F3, F5) but DNA extracted from these blocks was degraded and could not be amplified. DNA from the sampled patient in F2 was also degraded. DNA for segregation studies was obtained for parents and/or unaffected siblings from all seven families (F1 to F7).
The three cases which underwent exome sequencing are described. The first case was born at term. The baby was immediately brought to medical attention because of intestinal obstruction and omphalocoele. He died the following day of bowel obstruction. Autopsy revealed MIA. The second case is a fetus aborted at 23 weeks of gestation. Fetal ultrasound showed significant bowel distension at 22 weeks. Since the parents had a child who died at 2 days of MIA the previous year, a recurrence was highly suspected. Fetal autopsy showed MIA. The third case was born after a 35 weeks pregnancy, characterised by polyhydramnios. Intestinal obstruction was suspected at birth on clinical grounds. Imaging findings were consistent with a diagnosis of MIA. Considering the invariably fatal outcome of this condition in our centre, palliative care was given to the infant. Autopsy was declined by the parents and the baby died at home during the 1st week.
Patients with MIA have also been identified at the Centre Hospitalier Ste-Justine (CHU Ste-Justine) in Montreal.1 In one family (F7) two newly ascertained male siblings (P11 and P14), not previously described, were diagnosed with MIA. DNA was obtained from peripheral blood of one sibling, and from preserved tissue of the other. The family is reportedly of French-Canadian origin on the maternal side, though not known to be related to the CHUL and partial English ancestry on the paternal side.
The first sibling was born after 32 weeks of pregnancy complicated by polyhydramnios. Prenatal diagnosis of intestinal atresia was highly suspected because bowel distension was observed on fetal ultrasound at 25 weeks of gestation. Surgery performed at 2 days of life consisted in resection of multiple atresia involving the jejunum and the colon, and an ileostomy was created. Two subsequent surgeries were performed at days 16 and 35 to treat new atresias involving the pylorus and the jejunum. After resection, the patient presented bowel obstruction and died at day 47. The second sibling was born at 34 weeks after delivery was induced because of fetal distress. MIA was suspected based on echography performed at 22 weeks of pregnancy. During surgery performed at 1 day of life, multiple atresias involving the pylorus, ileum, proximal colon and rectum were resected. The progression of the disease required additional surgery at day 28. In spite of the treatment with steroids and cyclosporin A introduced at the age of 40 days because of a severe inflammation observed in biopsies, the intestinal disease progressed. The patient also had a severe immunodeficiency characterised by recurrent infections associated with hypogammaglobulinaemia and profound T cell lymphopenia. Haematopoietic stem cell transplantation was performed using a human leukocyte antigen (HLA) 6/6 matched cord blood at 6.5 months. The intestinal disease progressed and the patient died at 1 year of age.
Molecular studies
Informed written consent was obtained for genomic molecular studies using peripheral blood, immortalised cell lines or fixed tissue samples from deceased cases, and for studies of living first degree relatives. Genomic DNA was extracted from peripheral blood using the Puregene DNA isolation protocol. DNA was extracted from intestinal tissues embedded in paraffin or plastic using several rounds of phenol extraction and ethanol.
Exon regions were captured using the Agilent SureSelect All Exon 50 MB (V3) exome enrichment kit and sequencing of 100 bp paired end reads was carried out on Illumina HiSeq 2000. Capture and sequencing was performed at the McGill University and Genome Quebec Centre for Innovation. Sequence reads were aligned to the human reference genome hg19 using Burrows-Wheeler Alignment (BWA 0.5.9).11 Genome Analysis Toolkit (GATK) was used for realignment around suspected indels,12 and Picard was employed to mark duplicate reads and exclude them from downstream analyses.13 Single nucleotide variants (SNVs) and short insertions and deletions (indels) were called using Samtools mpileup.13 Mutations were annotated using Annovar14 and custom scripts. Filtering of common variants and recurrent technical artefacts of sequencing chemistry and/or read misalignment was performed using dbSNP132, Exome Variant Server (EVS), the 1000 Genomes variant dataset and a custom database of approximately 700 exomes previously sequenced at our centre for unrelated projects. Variants seen in more than 20 control exomes, SNVs with a ratio of variant reads less than 0.2 and insertion/deletions with a ratio of variant reads less than 0.15, were excluded from further analysis. Only variants predicted to change protein sequence (exonic non-synonymous SNVs, short indels, splice site SNVs) were considered. Private variants were defined as those absent from dbSNP, 1000 Genomes, Exome Variant Server and our internal exome databases. Variants were visualised using Integrative Genomics Viewer (IGV), or with NextGene (Soft Genetics, Inc.)
Sequences of primers for amplification and sequencing of TTC7A exons are given in online supplementary materials. Genomic DNA was extracted using the Puregene DNA isolation protocol from 28 ml of whole blood drawn by venipuncture. PCRs were performed on a Hybaid Omnigene Temperature Cycling System in a total volume of 50 ml containing 100 ng of genomic DNA, 20 pmol of each primer, 200 mM dNTPs, 50 mM KCl, 10 mM Tris (pH 9), 1.5 mM MgCl2, 0.01% gelatin, 0.1% Triton X-100 and 1 U Taq polymerase (Invitrogen, Burlington, ON). Amplifications were carried out using a ‘hot-start’ procedure. Taq polymerase was added after a 5 min denaturation step at 95°C. Samples were then processed through 35 cycles of denaturation (95°C for 30 s) and annealing (55°–60°C for 30 s), followed by one last step of elongation (72°C for 50 s). PCR products were diluted in five volumes of phosphate buffered (PB) buffer (Qiagen, Mississauga ON), transferred on a Whattman GF/C filter plate, washed twice with 80% ethanol/20 mM Tris (pH 7.5) and eluted in 50 ml of water. Samples were quantified by the PicoGreen reagent protocol. A second PCR was performed on Applied Biosystems Gene Amp PCR System 9700 (96 wells) or 9700 Viper (384 wells) machines to incorporate the sequencing dyes using a protocol of 25 cycles of denaturation (95°C for 10 s) and annealing (55°C for 5 s), followed by one last step of elongation (59°C for 2 min). PCR products were purified by the Applied Biosystems ethanol-EDTA precipitation protocol, collected using a Beckman-Coulter Allegra 6R centrifuge, and resuspended in a 50% HiDi-formamide solution. Samples were then run on Applied Biosystems Prism 3700xt DNA Analyzer automated sequencers. Sequence data were analysed using the Staden preGap4 and Gap4 programmes, or else using MutationSurveyor (Soft Genetics, Inc.)
Amniocytes were cultured from the exome-sequenced affected patient of pedigree 4 using standard culture procedures. For splicing analysis, RNA was extracted from cells and reverse transcription-PCR (RT-PCR) was performed using the Qiagen OneStep RT-PCR kit (Qiagen). The RT-PCR forward primer was designed in exon 4 with a few nucleotides extending in exon 5 to span the splice junction. Reverse primer was designed in exon 12 with a few nucleotides extending in exon 11 to span the splice junction (see online supplementary materials). This strategy allowed the screening of alternative splicing events in encompassed exons, and minimised amplification of genomic DNA.
Multiple sequence alignments were performed using ClustalOmega15 or Multiple Sequence Comparison by Log Expectation (MUSCLE),16 together with BoxShade. Putative TTC7A orthologs were obtained from the National Center for Biotechnology Information (NCBI) Gene and Homologene databases for the following species: Homo sapiens (modern human), Pan troglodytes (common chimpanzee), Macaca mulatta (rhesus monkey), Bos taurus (domestic cow), Canis lupus familiaris (domestic dog), Felis cattus (domestic cat), Equus caballus (horse), Loxodonta africana (African elephant), Oryctolagus cuniculus (European rabbit), Otolemur garnettii (northern greater galago), Mus musculus (house mouse), Rattus norvegicus (brown rat), Ailuropoda melanoleuca (giant panda), Monodelphis domestica (grey short-tailed opossum), Sarcophilus harrisii (Tasmanian devil), Meleagris gallopavo (wild turkey), Anolis carolinensis (green anole lizard), Danio rerio (zebrafish), Oreochromis niloticus (Nile tilapia). The putative ortholog of Gallus gallus (common chicken or red jungle fowl) is incomplete due to a problem with genomic annotation of the critical carboxy terminal coding exon and was omitted from alignments.
Missense mutation pathogenicity prediction was performed using PolyPhen2,17 Protein Variation Effect Analyzer (PROVEAN),18 MutationTaster,19 Sorting Intolerant from Tolerant (SIFT)20 ,21 and PMUT.22 Splice site mutation pathogenicity prediction was performed using MaxEntScan,23 ,24 and with the Berkeley neural network algorithm NNSplice, V.0.9.25 Because one putative mutation is intronic, DNA sequence numbering was based on annotated genomic contig NC_000002.11. Protein sequence numbering was based on RefSeq entry NP_065191.2.
Results
We first performed genome-wide homozygosity mapping using 408 microsatellite markers spaced at an average of 10 cm with DNA from three affected patients. As no obvious region was detected, we then sequenced the exomes of these patients from different families of the CHUL (F1, F4, F6, see figure 1). An average of 12.5 gigabases of raw sequence were acquired for each sample. After aligning reads to the consensus human genome using an implementation of the Burrowss-Wheeler algorithm (BWA), average mean read depth of bases in consensus coding sequence exons assessed by GATK was 91X. Average consensus coding sequence bases covered by at least 5, 10 and 20 reads were 94%, 91% and 86, respectively.
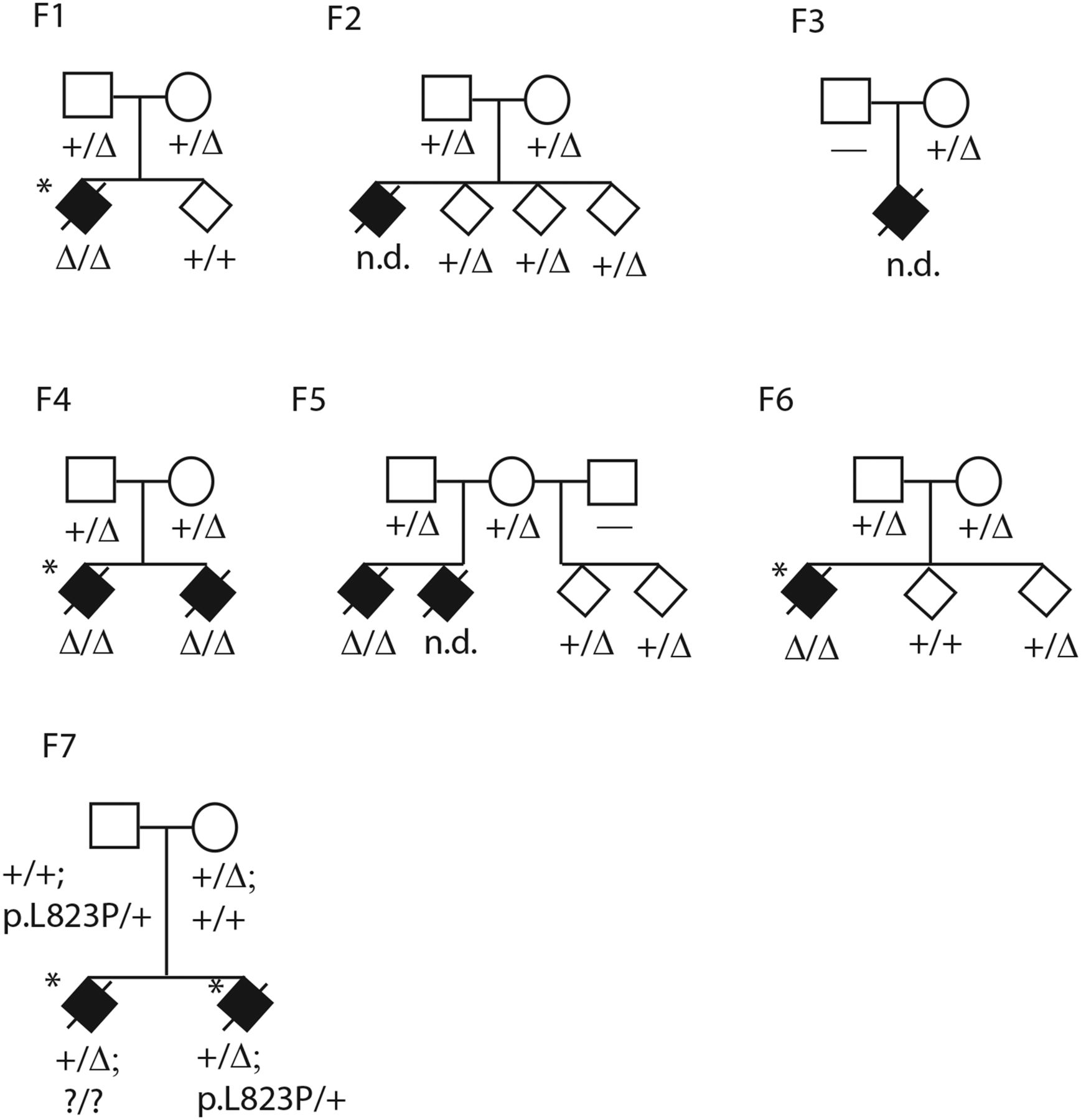
Multiple intestinal atresia pedigrees F1–F7. F1–F6 are from Centre Hospitalier de l'Université Laval cohort, F7 is from Centre Hospitalier Ste-Justine cohort. Affecteds are shown as filled diamonds, status of 4 bp deletion in exon 7 only is shown in F1–F6 as a delta sign, status of the 4 bp exon 7 deletion and p.L823P missense in exon 20 are shown in F7. Horizontal dash or question mark means no genotype was obtained. Probands who underwent exome sequencing are starred. One affected was previously reported (Bilodeau et al, case 11).2 To preserve confidentiality sex is not shown, and children are not in obligate order of birth.
As in other published studies, each individual exome sample contained more than 100 000 called variants. After filtering with public and custom exome variant databases to remove common SNVs and recurrent technical artefacts, each sample contained about 400 rare non-synonymous variants (predicted to alter protein function or splicing). Only one variant was shared in the homozygous state among all three patient exomes, this was a four base pair deletion c.53344_53347delAAGT in the gene TTC7A, encoding TPR protein 7A (MIM[609332], figure 1, see online supplementary figures S1, S3A). No gene contained two different pathogenic variants (potential compound heterozygotes) in all three patient exomes. The 4 bp deletion occurs at the 5′ splice donor site of exon 7, where the consensus genome sequence contains a directly repeated AAGT forming the GT splice donor site. The deleted allele contains only one AAGT. The variant falls in a region of extended homozygosity of several megabases in each of the three patients based on exome-derived high quality variant calls. The exact length of homozygosity varies among the three patients, while the minimal overlapping shared region of homozygosity was approximately 1.2 Mb. For the most part all three affecteds share minor SNV alleles identical by state across this region, although exome software is not optimised for such an analysis.
To validate the homozygous AAGT deletion, all 20 coding exons of the TTC7A gene and their intron-exon junctions were screened for variations by PCR-based Sanger sequencing in the three exome sequenced samples and available members of their nuclear families (F1, F4, F6). The only variation detected in all three patients was the 4 bp homozygous deletion. The other 19 coding exons had no interesting sequence variants. In all three families, both parents were heterozygous for the deletion and unaffected siblings were either heterozygous for the deletion or homozygous wild type (figure 1). In family F2, DNA of the affected deceased case was unavailable but both parents were heterozygous for the deletion and unaffected siblings were heterozygous for the deletion (figure 1). In family F3, one available parent was heterozygous for the deletion. DNA from two additional MIA patients (families F4 sibling of exome sequenced patient, F5 one of two affected patients) were extracted from embedded intestinal tissue biopsies. Both these patients also carried the homozygous AAGT deletion, and parents of the F5 patient were both heterozygous for the deletion.
Although the deleted allele retains a potential GT splice donor site, the mutation is predicted to have a deleterious effect on splicing, with a maximum entropy score of −9.7 according to MaxEntScan. Similarly, the Berkeley splice site prediction programme NNSplice correctly identified the splice donor site at the distal end of exon 7 using 132 bp of wild type genomic sequence bracketing the site, but did not predict any donor site in the same region with the 4 bp deletion. To assess potential aberrant splicing, poly-A+ RNA was extracted from Hela cells or from cultured amniocytic cells of one patient (from pedigree 4) and from one normal control infant. Following RT-PCR, TTC7A RNA was detected from the patient cells, as well as from control and Hela cells. Consistent with the bioinformatics predictions, a smaller size product was observed from RNA of the patient, versus RNA from control or Hela cells, for PCR reactions of cDNA spanning exon 7 (see online supplementary figure S2). Sequencing of the smaller product verified that exon 7 was skipped, with splicing of exon 6 directly to exon 8 (see online supplementary figure S3B). This aberrant splicing generated a 158 bp deletion in the resulting cDNA, predicted to cause a frameshift, 281 amino acids through the gene with 49 new amino acids followed by a termination codon, with a presumptive severe effect on protein function (full length wild type sequence is 858 amino acids). In human lymphocyte RNAseq data from other unrelated projects, we examined the state of TTC7A splicing and found rare examples of reads skipping exon 7 (four reads excluding exon 7 vs 880 reads including the exon), suggesting that exon 7 inclusion may be intrinsically variable even with a wild type splice donor site.
An additional family (F7) with two MIA affected siblings was independently ascertained at CHU Ste-Justine. Exome sequencing identified the same 4 bp AAGT deletion seen homozygous in the CHUL, but in the heterozygous state in both siblings. The entire TTC7A gene could not be interpreted due to variable exon sequence coverage. All 20 TTC7A coding exons and their intron-exon junctions were therefore screened for variations by Sanger sequencing, confirming that the two siblings were both heterozygous for the 4 bp AAGT deletion (figure 1, see online supplementary figure S4). A second heterozygous mutation was found in both siblings: c.A133074G; p.L823P, in carboxy-terminal coding exon 20 of the gene (figure 1, see online supplementary figure S3C). By Sanger sequencing, each parent was heterozygous for one of the two mutations, the 4 bp deletion on the maternal side consistent with her French-Canadian origin, and the missense on the paternal side. Together the results are consistent with the mutations being compound heterozygous in the two affected patients of this family, although we were unable to test the exon 20 mutation in the other affected sibling directly. The leucine at position 823 in the human gene sequence is highly conserved among putative TTC7A orthologs (figure 2), and mutation p.L823P is predicted as probably damaging by PolyPhen2 HumDiv and HumVar, as deleterious by PROVEAN (score −5.7, anything lower than −2.5 is considered deleterious), as damaging by SIFT, as disease causing by MutationTaster and as pathological by PMUT. In the TTC7A gene itself, SNPs identified as homozygous in the three homozygous affecteds were heterozygous in the two affecteds of the compound heterozygotes, or else had low overall read coverage.

{kind=link}
{kind=link}
Multiple sequence alignment of putative TTC7A orthologs, showing the C-terminus of the protein. Residue L823 is boxed in all orthologs, and the mutation p.L823P (according to the human amino acid numbering) is shown above the top row.
Neither the exon 7 AAGT deletion nor the exon 20 missense mutation in TTC7A is found in dbSNP V. 135 (which includes data from the 1000 Genomes project), and neither was observed in 1500 French-Canadian control chromosomes (although as noted the paternal missense mutation may be of British or Scottish origin).26
Based on our molecular genetic results, TTC7A is a strong candidate for the MIA phenotype in all our analysed cases.
Discussion
Exome and PCR-based sequencing studies of seven cases of MIA, as well as obligate carriers and unaffected relatives from seven families, all from French-Canadian Quebec, identified two different mutations segregating in the gene encoding TTC7A. One mutation, a 4 bp deletion of one copy of a duplicated tetranucleotide encompassing a splice donor site, was predicted to be pathogenic by two different splice prediction tools. Five of the cases were homozygous for this mutation, which was also found in the heterozygous state all available parents including those of additional affecteds whose DNA was not directly available. The variant was not seen in the homozygous state in any unaffected family members of probands. This deletion has also not been seen in any other exome sequence data in our laboratories, which include a large number of French-Canadian samples. In direct RT-PCR experiments, the aberrant splicing product was observed in cultured cells from one of the patients, consistent with the bioinformatics predictions. Skipping exon 7 leads to a predicted severe protein truncation. Although the aberrantly spliced product should be susceptible to nonsense-mediated decay, sufficient RNA was nonetheless present in the patient's cells to permit the assay. The second mutation, missense p.L823P, occurs at a highly conserved residue near the carboxy terminus of the protein, and is predicted to be pathogenic by five different programmes. Two cases, siblings in the same family, were likely compound heterozygotes for the 4 bp intronic splice site deletion and the missense mutation (second hit verified in one sibling). These results, together with SNP calls derived from the exome data (not extensively revalidated), are consistent with TTC7A mutations being causal for the intestinal atresia in all of these cases. It is likely that the 4 bp deletion represents a founder mutation in the French-Canadian population, with the missense mutation having arisen more recently or else derived from a British or Scottish population; similar observations of combined founder and non-founder mutations in the same gene have been observed for other monogenic phenotypes in eastern Canada.27 It remains to be seen whether all ascertained cases of congenital MIA are caused by mutations in TTC7A or whether additional genetic or non-genetic aetiologies are also significant.
Little is known concerning the function of TTC7A in human development. The TPR domain consists of approximately 34 amino acids with a consensus sequence of (WLF)-X(2)-(LIM)-(GAS)-X(2)-(YLF)-X(8)-(ASE)-X(3)-(FYL)-X(2)-(ASL)-X(4)-(PKE) according to the NCBI conserved domain database.28 This appears to be a protein-protein interaction domain with various potential partners involved in different cellular processes including cell cycle control, phosphate turnover and subcellular localisation,29 ,30 thus it does not suggest any particular physiological role for TTC7A itself. Multiple TPR domains occur at the carboxy-terminal end of TTC7A, and the missense putative mutation p.L823P is in one of these. Physical studies indicate that TPR domains possess significant α-helical structure,29 hence since proline residues are strong helix-breakers the L-to-P mutation is likely to disrupt the structure of that TPR repeat. TTC7A appears widely expressed at low levels, though at slightly higher levels in myeloid and monocyte cells according to the Bio Gene Portal System (BioGPS) human tissue expression database.31
Interestingly, three spontaneously arising mutations in the mouse TTC7A ortholog, Ttc7, are known. The flaky skin (fsn) recessive mouse was originally described in 1995, with a phenotype including a papulosquamous disease of the skin, anaemia with an associated haematopoietic disorder and gastric forestomach hyperplasia.10 Subsequently another spontaneous recessive mouse mutant, hereditary erythroblastic anaemia (hea) was found and determined to be allelic to fsn.9 The hea strain shares the haematopoietic and skin aspects of the phenotype,32 but not reportedly the stomach hyperplasia,9 although the skin phenotype was only observed when the mutation was transferred from the original CFO genetic background to C57Bl/6J.9 The common causal gene for fsn and hea was positionally cloned as Ttc7, the mouse ortholog of human TTC7A.33 The hea allele results from a large deletion encompassing exons 1–14, whereas the fsn allele results from an intronic transposon insertion that leads to incorporation of a transposon-derived extra exon between mouse coding exons 14 and 15; this leads to an inframe insertion of 61 amino acids into the protein disrupting one of the TPR domain repeats.33 ,34 In terms of phenotypic pleiotropy the fsn allele seems slightly more severe, although the hea mice reportedly have shorter life spans. The fsn mice provide an important model system for human psoriasis, and it is unclear whether the anaemia and associated iron deficiency is a primary or secondary aspect of the mouse mutations. The fsn mice have abnormal lymphoid cell populations including a reduced ratio of CD4 to CD8 cells and massively enlarged peripheral lymph nodes, moreover lymphoid cells are hyper-responsive to interleukin growth factors.35 A third allelic mouse mutation in the gene was later identified, int, and shown to result from deletion of exons 9–10, leading to an inframe protein deletion.8 ,36 The int mice have severe haematopoietic and papulosquamous skin disorders and die as neonates, however they do not appear to exhibit a gastrointestinal phenotype.
These mouse mutants exhibit quite different clinical presentations from human patients. The mouse genetics do indicate that Ttc7 plays a role in proper immune system function, which is consistent with an immune involvement in MIA. However, the immune phenotype of the mice is different from what is observed in human patients, who present with a SCID-like phenotype in most of the cases in which an immunodeficiency is observed. In addition, the mice have no documented intestinal involvement (although the fsn strain does have forestomach papillomas), whereas the human patients have no obvious skin involvement. Conceivably these differences are due to allele-specific effects on immune function; perhaps the human mutations are hypomorphic compared to the mouse mutations. The lack of intestinal involvement in the mice especially in the large deletion strain is difficult to reconcile, although the much shorter intestinal tract and differences in development of intestinal mucosa and immune defence in rodents may be relevant. Alternatively, even the severe mouse mutations show significant genetic background effects, thus modifiers elsewhere in the genome may be relevant to the human phenotype of MIA. Treatment of fsn mice with helminth (worm)-derived glycans ameliorates the skin phenotype.37 Helminth extracts have been tested in humans for other autoimmune disorders with some promising results.38 Given the typically fatal course of MIA and lack of any therapeutic regime other than pre-emptive surgery to remove damaged tissue, it is worth speculating whether glycan treatments either prenatally or postnatally might be of benefit. Finally, although stem cell transplantation during the 1st year of life for the SCID was unsuccessful in one patient, conceivably fetal stem cell transplantation through the fetal cord might represent a therapeutic option, if indeed the intestinal phenotype is secondary to an immune system dysfunction. This however remains to be established.
Acknowledgments
Foremost we thank the families who generously contributed their time and materials to this research study. Thanks to Maryse Lagace, Lysanne Patry, Renée Dicaire and Christine Massicotte for assistance with patient and sample coordination and ethics submission. The FORGE Canada Consortium Steering Committee consists of Kym Boycott (leader; Ottawa, Ontario), Jan Friedman (co-leader; Vancouver, British Columbia), Jacques Michaud (co-leader; Montreal, Quebec), Francois Bernier (Calgary, Alberta), Michael Brudno (Toronto, Ontario), Bridget Fernandez (St Johns, Newfoundland), Bartha Knoppers (Montreal, Quebec), Mark Samuels (Montreal, Quebec), Steve Scherer (Toronto, Ontario). We also thank Janet Marcadier (Clinical Coordinator) and Chandree Beaulieu (Project Manager) for their contribution to the infrastructure of the FORGE Canada Consortium.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors AH participated in bioinformatic analyses of whole exome data, BM was responsible for clinical ascertainment and phenotype definition, and participated in manuscript preparation, EH participated in clinical ascertainment and phenotype definition and in manuscript preparation, FC participated in exome sequencing, molecular studies and data analysis, FLD was responsible for clinical ascertainment and phenotype definition and participated in manuscript preparation, HD participated in clinical ascertainment and phenotyping patients, IF participated in phenotype definition and manuscript preparation, IG assisted with clinical ascertainment, phenotype definition, patient sampling and ethics approvals, JM assisted in clinical ascertainment and manuscript development and participated in manuscript preparation, JLM coordinated bioinformatic analysis of whole exome sequence data and participated in manuscript preparation, MAR participated in bioinformatic analyses of whole exome data and in molecular studies, MES coordinated molecular studies and primary manuscript development, NA participated in bioinformatic analyses of whole exome data, NP participated in phenotyping patients, VR supervised molecular validation and RNA-based studies and participated in manuscript preparation, PA coordinated bioinformatic analysis of whole exome sequence data and participated in manuscript preparation, SyD participated in molecular studies, StD participated in molecular studies, VM participated in clinical ascertainment and phenotyping patients, YI participated in exome sequencing, molecular studies and data analysis.
-
Funding This work was funded by the Government of Canada through the Canadian Institutes of Health Research, Genome Canada, Genome Quebec, Genome British Columbia and the Ontario Genomics Institute (OGI-049). MES was supported by the Centre de Recherche du CHU Ste-Justine. PA, FLD and EH were supported by CIHR Grant 120299. EH is a scholar of the Fonds de recherche du Quebec-Sante. VR was supported by Fonds de recherche du Québec- Santé.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was approved by the research ethics committees of CHU Ste-Justine and CHUL.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Data from this study may be shared with other researchers.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/